-New things are placed here continuously-
7.) 1H+13C NMR + 13C-DEPT Spectra of DMT with max purity
derived directly from Extraction (No Re-X)
This time I wanted to unravel
how pure Spice could get - directly derived from the Non-Polar-Solvent, meaning that there is
not a single purification step after the Spice was obtained in its solid form.
The solvent used was a mixture of isomeres of
Hexan + Pentan, meaning that this kind of Naphtha is the lowest boiling you could get (= 40 - 60 °C as opposed to 60 - 80 °C with Hexan + Heptane), which pulls the least other junk things besides the Spice. Then it was frozen-precipitated to give exactly what was measured here. Still overall solubility is much lower, therefore I dont recommend using it.
Sample:
Here is the 1H-NMR of that sample:
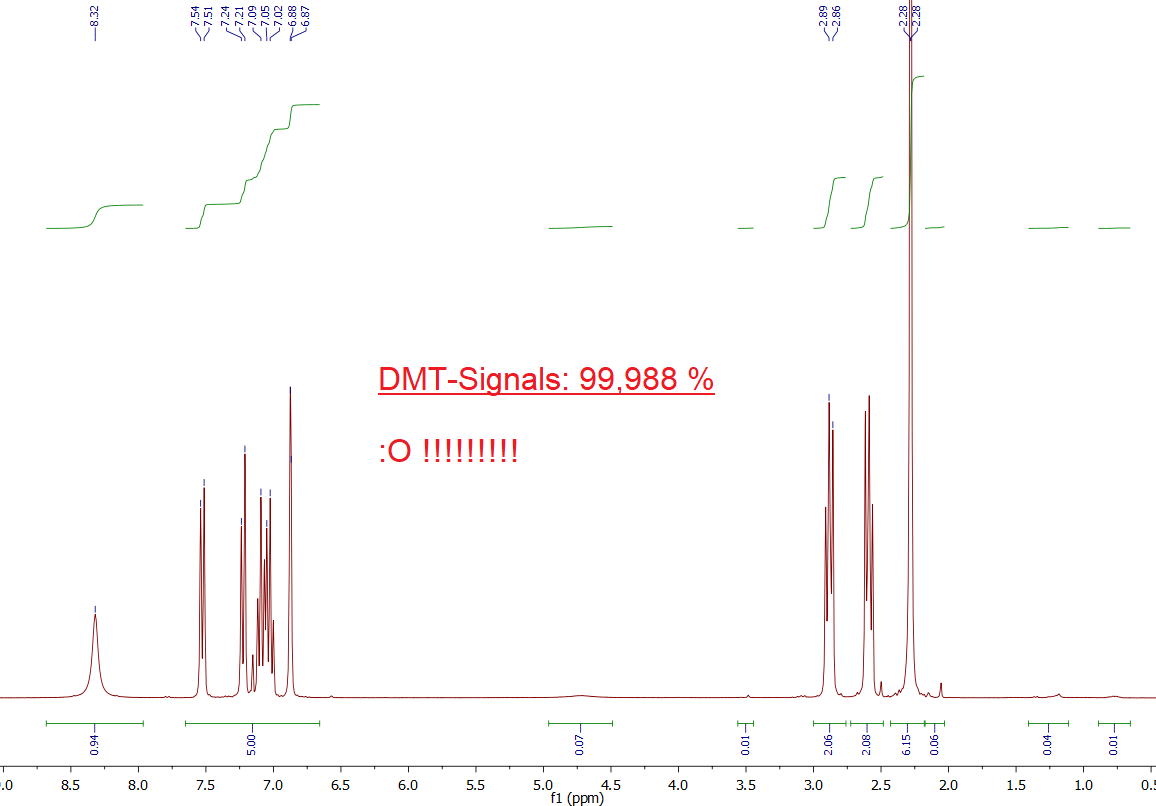
Observation:
Well to make it short: The spectrum is PERFECT. It contains - as always - all relevant DMT Signals - but it has NO other signals. Or at least those other signals are so tiny, you may really say that this even looks like the *Predicted Spectra* in Chapter
1.). This is really astonishing, as no workup method was used and this sample was derived directly from the freeze precipitation. Seems like in some cases it is not even possible to perform any workup, as the product is already practically 100 % DMT-only.
Here is the 13C-NMR of that sample:
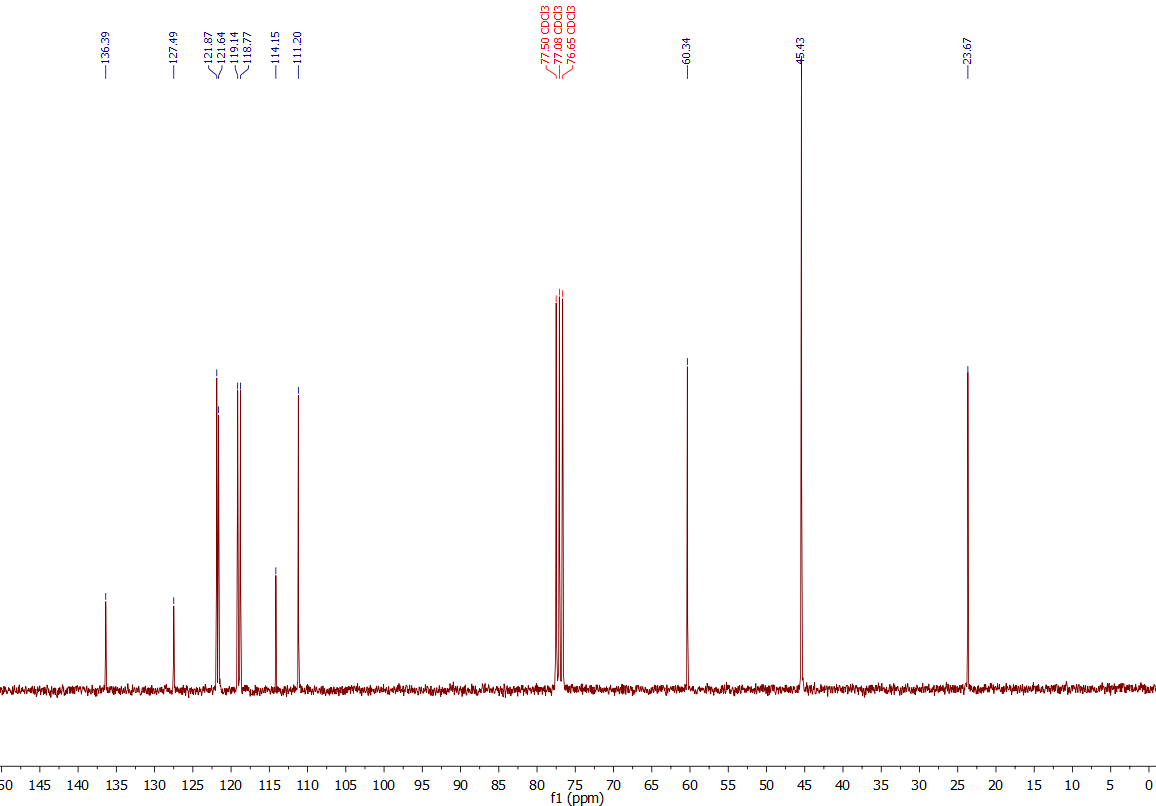
Observation:
Same case here: Again it shows only 100 % the signals of DMT - nice!
EDIT: So now here just for the completion as I finally have a let's say *Pure Sample* there is the "
Distortionless Enhancement by Polarization Transfer" Spectrum -> *13C-DEPT NMR*
What is this? An additional key to unravel an unknown structure of a compound by telling what
carbon atoms - based on their respective C-NMR Signals - carry
how many protons or if they carry them at all. As we already know the structure of our sample of course, this is really irrelevant, but mostly just for the completion to present you a full set of NMR data of basically pure Spice.
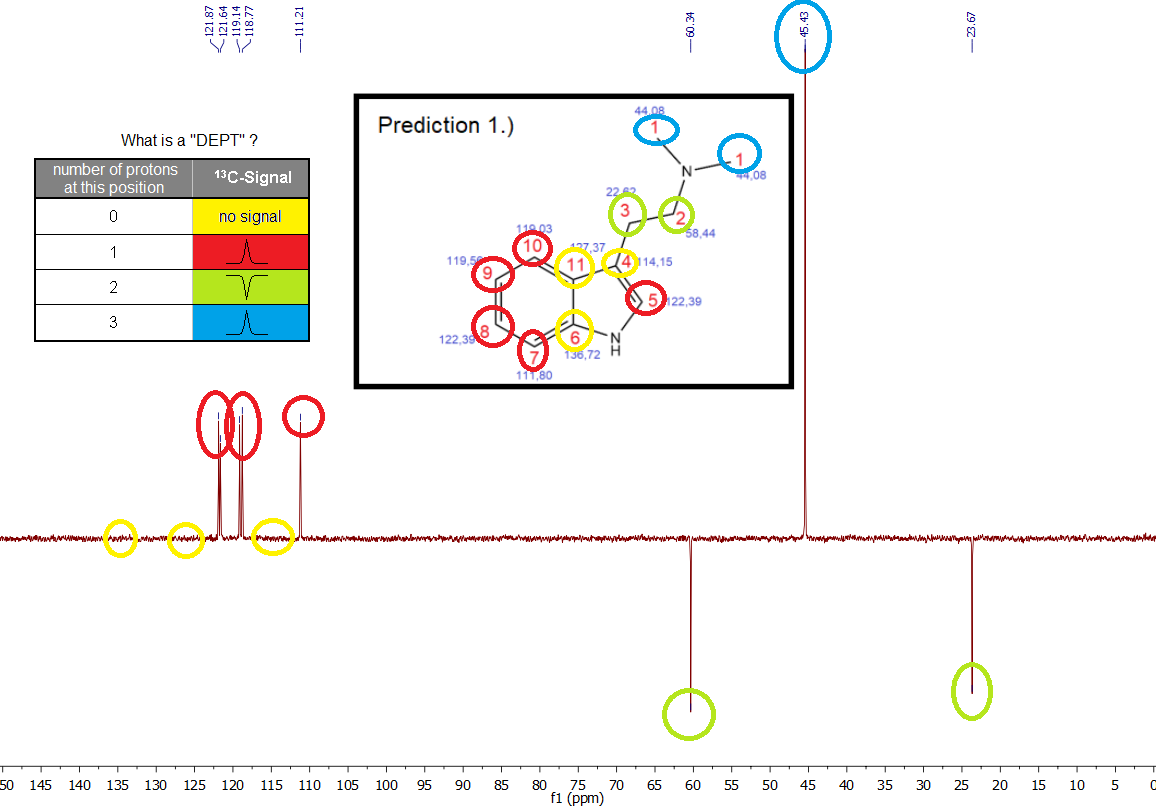
Yes ... not that interesting, you just see what you expected. This DEPT method will be much more interesting when we have the impurity sample.
Conclusion:
Well this seems that when you use the lowest-boiling Naphtha (40 - 60 °C, Hexane +
Pentane) and
PROPERLY wash the Non-Polar-Solvent in the end with a sodium (bi)carb-wash or something comparable and freeze-precipitate it afterwards you can already get
practically 100 % pure Spice.
Normally in the science about plant ingredients and chemicals its a pain to get pure products or to verify a possible way to isolate them in a mostly pure form.
It seems that it is just a gift of heaven that Spice can be isolated that perfectly by that *easy* TEKs that are floating around here.
Based on this I would say that if your Spice is totally white and you did the regular steps until the freeze-precipitation, then there is totally no Re-X worthy - I even told in the upper Chapters that even yellow Spice seems pretty good - but in case you really want totally pure Spice, then it is definetly not worth to Re-X if it is already clear white.
Of course there COULD be some other components in your mixture that are also white, which were not present in this sample here. But I guess it should be highly unlikely that there may be any other compound that could cause any potantial relevant contamination, if it is not visible here even in a trace amount.
Additional analysis stuff of pure Spice
For the sake of complicity, here is the UV-Vis spectrum of DMT. Actually I have no idea, why anyone would need / use that, but I have it so here you go. If you look for more general chemical data on DMT and many other molecules, better just directly see
here.
DMT in Acetonitrile, c = 7,5*E-4 mol/l
It's quite boring and no absorption in Vis-area, but hey, thats why we get white Crystals.
Also for complicity, here is the IR-spectrum of DMT. Same as above, but maybe it may prove useful somewhere in future. As it takes some time to analyse an IR-spectrum (at least if its not your personal hobby) there are no further details. Measured in attenuated total reflection mode.
What is also missing is an XRD of DMT - its an analysis on the crystalinity. That would be quite interesting, as it may get a deeper insight into the different DMT morphologies, that also have individual melting points. That may highly vary based on crystalinity, which an XRD can observe. I had the opportunity to use one in the past, but I did not have the balls. Maybe I may get an XRD of different morphology samples in the future.
8.) 1H-NMR of Harmala Alkaloids (No Manske)
This is just an 1H-NMR taken from a crude Harmala Alkaloid mixture. Harmalas were extracted with acid and simply precipitated with NaOH and washed until pH was neutral. No Manske precipitation was done, I have the feeling that it takes ages for these crystals to sink and filtering is not easy ...
Sample:
Here is the 1H-NMR of that sample (in DMSO):
Observation:
Not much to say here. Basically all the signals that can be definetly aligned to just 1 of either Harmin or Harmalin
show a pretty good 1:1 of both compounds. Wikipedia says this:
Harmin: 0,44 %–1,84 %–4,3 %
Harmalin: 0,25 %–0,79 %–5,6 %
Vasicin (Peganin): 0,25 %
(Harmalol could be there, but will be hidden below other signals)
So also Wikipedia mostly says it's roughly the same, but in my case that sample really is pretty much a plain 1:1 with a difference of at highest 9 % more Harmalin (Methyl-Peak ~ 2,3/2,5 ppm). Now can there any other information be retrieved? Harmalol-only signals would all be hidden below what we currently see. But Vasicin could be identified: It shows up with Signals at:
4,5 ppm (s) (CH2 between aromat and N)
3,8 ppm (m) (CH at Hydroxyl-group)
3,4 ppm (m) (CH2 at alpha-position in Pyrrolidin-ring)
1,7 ppm (d,m) (CH2 at beta-position in Pyrrolidin-ring)
Now only 4,5 ppm will be not hidden in that spectrum and we can see no peak. So I would say quite confidently that there is
absolutely no Vasicin in that sample. There was totally no workup step, so the Alkaloids were just crashed from the acidic boil. It is written that Manske-precipitation is needed to get rid of Vasicin. But actually it is also reported that there is no need for that, other than you would be pregnant. So I never made that hassle - but here obviously there is not even Vasicin present. So as it has a Hydroxyl-group, the solubility in water must be slightly higher than from the other 2 Alks. And as these samples were washed with excessive water, it must have been removed - a few g of Alkaloids were washed with 20 - 30 L of water.
So I may say: Just stop the Manske precipitation, which takes ages and may just make you loose product ... Instead put your freshly NaOH-precipitated Alkaloids from acidic boil in a 5 L plastic bottle and fill it with water, shake shortly and let it settle. This takes less than 5 min. Then decant and repeat. Do this until the water is clear and/or pH = 7 is reached.
This step will effectively combine:
1. Neutralization of any residual NaOH (so mandatory anyways)
2. (
possibly) removal of any Vasicin. Takes no time compared to Manske precipitation!
Here is some information from _Trip_ about using Ethyl Acetate as an extraction solvent, which will also not carry over Vasicin when doing a citrate precipitation later on:
_Trip_ wrote:I would like to add EA may not pick up vasicine even when using the EA dry tek approach. Although it could be more likely it doesn't form a citrate.