DMT-Nexus member
Posts: 465 Joined: 18-Jan-2008 Last visit: 09-Nov-2025
|
A compilation of my work with Phalaris. mistakes were made
|
|
|
|
|

DMT-Nexus member

Posts: 1760 Joined: 28-May-2009 Last visit: 10-Oct-2024
|
I just read through your notes and experience at the end and I have to say WOW! I am so impressed with the work you've done. Really invaluable info there. Phalaris is such a readily available plant all over the world, so the more successful work done on it, the better. Thanks for all the effort you must have put into this, fourthrpley.
|
|
|
analytical chemist
  
Posts: 7463 Joined: 21-May-2008 Last visit: 09-Aug-2025 Location: the lab
|
very nice account indeed. I did a sonication on some phalaris a while back, followed by a quick A/B. the product looked identical after re-x. Brachystachys is certainly the way to go, yield-wise. "Nothing is true, everything is permitted." ~ hassan i sabbah
"Experiments are the only means of attaining knowledge at our disposal. The rest is poetry, imagination." -Max Planck
|
|
|

DMT-Nexus member
Posts: 472 Joined: 19-Mar-2009 Last visit: 22-May-2023
|
This is a very valuable PDF, thanks for producing it. Everything I post is made up fiction. SWIM represents a character who is not based in or on reality.
|
|
|

DMT-Nexus member
Posts: 1711 Joined: 03-Oct-2011 Last visit: 20-Apr-2021
|
Excellent, very informative. The push I needed to seriously consider getting phalaris seeds. Thank you very much. "The Menu is Not The Meal." - Alan Watts
|
|
|

DMT-Nexus member
Posts: 14191 Joined: 19-Feb-2008 Last visit: 20-Jun-2026 Location: Jungle
|
Good work, thanks a lot  One question, why do you affirm with so much certainty about n-oxide? On what was your identification based, on the fact that it was a yellow oil only? There could be other explanations for it that dont involve n-oxide.. Also, did you do a 'control group' extraction (redissolving and adding base/extracting but without zinc), to compare with your supposed reduction? If you just add zinc to some vinegar (slightly yellow) even without any alkaloids, it will turn clear after a while, so the color change isnt necessarily an indication of reaction with the alkaloids. AFAIK dmt n-oxide was never found in phalaris, no indication of it with TLC (neither in my own tests) and neither with HPLC tests (like festi and samorini)... There is the possibility it formed during extraction (or that its there in some phalaris that just werent tested) but again, maybe it isnt, and it could be other things like plant oils that prevent dmt from crystallizing, and/or small solvent traces that also prevent it from crystallizing, etc..
|
|
|

Chalgren
Posts: 225 Joined: 14-Sep-2011 Last visit: 23-Aug-2014 Location: Limbus
|
Just wow, excellent work, effort well worth! Mad, bad and dangerous to know.
There's magic out there!
|
|
|

DMT-Nexus member

Posts: 12340 Joined: 12-Nov-2008 Last visit: 02-Apr-2023 Location: pacific
|
wow thank you for this! I am so glad others have been working with the grasses so we can figure this out. Long live the unwoke.
|
|
|

DMT-Nexus member
Posts: 793 Joined: 23-Oct-2011 Last visit: 22-Aug-2014 Location: arcady
|
I can only imagine this document inspiring more and more good stuff. Thank you. "Whoever undertakes to set himself up as a judge of Truth and Knowledge is shipwrecked by the laughter of the gods." Albert Einstein
I appreciate your perspective.
|
|
|
DMT-Nexus member
Posts: 12340 Joined: 12-Nov-2008 Last visit: 02-Apr-2023 Location: pacific
|
benzyme wrote:very nice account indeed.
I did a sonication on some phalaris a while back, followed by a quick A/B. the product looked identical after re-x. Brachystachys is certainly the way to go, yield-wise. So did you bioassay it? Long live the unwoke.
|
|
|
DMT-Nexus member
Posts: 12340 Joined: 12-Nov-2008 Last visit: 02-Apr-2023 Location: pacific
|
fourthripley, did you every bioassay the arundinacea extracts? My own experience with extracting and testing(on myself) wild arundinacea was similar to what you described of your experience with brachy's..only I never went quite as far as you did. I tested lighter doses a few times and there was definatily tryptamine effects pesent..Only once did I smoke a bit too much and this was very much like what I have read of 5meoDMT..it was frightening but facinating. I have alot of experience with DMT but none with pure 5meoDMT though so I cant be 100% sure what it was that caused that effect. Interesting that you find an increase in alkaloids in grass harvested in the fall..that has been my observation as well with wild grass. I must say that these reports of grass extractions that include bioassays are worth so much..too often a thread comes up with some extraction pics etc..someone extracts a yellow goo or something and then the thread just dies..no bioassay..which just sort leaves us back where we started. We all know grass can yield a yellow goo..but analysis and bioassay are what we need. Long live the unwoke.
|
|
|
DMT-Nexus member
Posts: 465 Joined: 18-Jan-2008 Last visit: 09-Nov-2025
|
endlessness wrote:Good work, thanks a lot Also, did you do a 'control group' extraction (redissolving and adding base/extracting but without zinc), to compare with your supposed reduction? If you just add zinc to some vinegar (slightly yellow) even without any alkaloids, it will turn clear after a while, so the color change isnt necessarily an indication of reaction with the alkaloids. AFAIK dmt n-oxide was never found in phalaris, no indication of it with TLC (neither in my own tests) and neither with HPLC tests (like festi and samorini)... There is the possibility it formed during extraction (or that its there in some phalaris that just werent tested) but again, maybe it isnt, and it could be other things like plant oils that prevent dmt from crystallizing, and/or small solvent traces that also prevent it from crystallizing, etc.. Endlessness; of course without access to the relevant tests etc. my identification of N oxide is of course speculative. However, I came to this tentative conclusion on the basis of several observations made during the process which I shall detail here. Hope I can remember and organise them First off, the smell. First results were almost discarded because the aroma although obviously related to, has a markedly different quality to Hostiolis extracted NN. It is sweet and extremely pungent, dishes of dried yellow oil must be double film wrapped so as to not fill the entire house. On vaporisation however the material smells identical to Hostilis freebase. After zinc reduction the recovered material smells identical to Hostillis extracts. Initial bioassay led me to believe the extract had a high level of purity. I assumed it should only be a matter of a few steps to recover crystals. Some pulls appeared to be highly saturated yet placing in the freezer resulted in no precipitation. I attempted simple recrystalisation with no success and re-a/b attempts with defats- trying both naphtha and DCM- mate no difference. The evaporated deffatting solvents revealed negligible amounts of oils remeved. I became somewhat despondent as to whether the material could be crystalised at all. I made an interesting observation: material was dissolved in acetone and set to evporate in a metal dish. Of course as the acetone evaporated it chilled the dish causing H2O to condense around the edges of the bowl. I observed the material actually chasing up the sides of the bowl towards the condensation, leading me to believe that some of the material was actually somewhat polar- as N oxide is said to be- and hydrophillic(?). I have never noticed this 'chasing' with samples known to be fb dissolved in acetone. Stability: I have noticed both NN and 5meo fb to be rather stable. I have material stored not particuarly well that appears to be as good as the day I put it away. The yellow oil appears to lose potency over the course of a year. As stated in the document year old samples, which had been bioassayed and shown to be powerfully active, after zinc reduction and re a/b yielded nothing. No oils or junk either. It is recorded that Phalaris loses potency on drying. My extractions on dried material have yielded next to nothing. I ran straight a/b's on similar quantities to the one I talk about. All yielded little more than smears. Either the material is not picked up in a standard a/b or is somewhat destroyed during the process. I include an early extraction note from 2008, when I was starting to explore the plant, with anomalous findings and my naive musings at the time as to what could be going on: Quote:#1: 300g of fresh p.b whole plant was wrung through a wheat grass juicer to yield maybe 300 mls of juice. This was brought quickly to a boil- the reasoning being to denature the enzymes said to cause degradation of alkaloids in Phalaris sp. - the liquid appeared to separate out into a brown, thin liquid with some sedimentation floating at top and bottom. The whole thing was passed through a polyfill plug in a funnel to yield a fairly clear brown liquid and a vivid green sedimentation in the funnel. The green material was washed out of filter and dried. The brown liquid was heated and 7 defats were performed with boiling mineral spirits, yielding fair amounts of white looking goo which floated in the yellow tinged solvent. At 7 there appeared to be little gunk pulled. The aqueous solution was basified with 50g NaOH and extracted with 50mls hot naphtha 3 xs. The first pull was evappped to leave a trace of yellow oil which from its smell would appear to be NN or a near relative. The combined second and third pulls yielded nothing. The goo was separated from the defatting solvent and evapped. I was surprised to find that as it dried the goo collapsed into an orange powdery material, maybe 20 mg, when fully dried I scraped up the orange material and very casually mixed it into a small tobacco cigarette. Expecting nothing more than chest hurt and a desire to extinguish, I was taken aback to experience euphoria and stimulation of a Shulgin level one level and a strong desire to finish the smoke. The room filled with the lovely aroma of fresh spice. Really making no sense to me at all. The alkaloid exists in the plant as fb? The alkaloid salts are some how caught up in the plant fats in some sort of weird ass emulsion? Of course N oxide is said to be soluble in pet ether containing fats. Mineral spirits are essentially heavy pet ether or thereabouts. Perhaps that's what was caught in the defatting solvent. On acidifying the material the solution is opaque, more like a colloid than a soltion; after the addition of zinc it quickly becomes transparent, preceding any colour change. Finally, from Trouts notes a fleeting reference in the following abstact- Woods and Clark (1971) Crop Science 11-121-122: Quote:...The aqueous solution was then extracted with chloroform which was taken to dryness to yield a residue containing crude alkaloids.
Aqueous solution of DMT-N-oxide/ acidify with acetic acid/ reduce with excess of zinc dust/ stir 30 minutes/ raise to ph 9 with NH4OH/ extract three times with chloroform. mistakes were made
|
|
|

Faustian Phytochem Investigator
Posts: 194 Joined: 31-Oct-2011 Last visit: 29-Nov-2025 Location: Oaxaca
|
Thanks for posting this, I think there is some really good information here. It's going to be hard to suss out anything definitive, but it really points in some good directions. As for the oil being an oxide, IMO its a possibility. The smell seems consistent (tho I would describe it as floral and sweet, not so much pungent). If this is the case, I would be more inclined to consider the conversion of the amines to their oxides via the procedure, IE: an artifact of the extraction. I base this off of the reading of articles dealing with the oxides in extractions stating that they do form as artifacts. As for the Zn reduction: as endless points out here, color alone is not a good indicator for anything other than *some* change has occurred. Without running at least TLC on the samples, it is almost impossible to say what this change is. You can run this reaction on MHRB with similar color changes, and this seems to be affecting the tannins in the extraction as they will precipitate out (heavily); but this is not due to DMT-oxide it is the reaction happening to another compound. Also from your paper: Quote:To counter this, after Zn reduction, I perform 1 DCM defat, obviously checking the ph of the solution is low
enough. While this would definitely help things to crystallize, it throws another variable into the mix that complicates things. My thinking here is that by performing this step, you will not necessarily know if it was any oils in the material or the oil of the oxide that was preventing crystallization, you've effectively dealt with both at once. Side by side comparisons of the same material would lend more evidence to one or the other possibilities. That is, do one with only the reduction, do one with only the defat, and one that incorporates both. Then compare results. Thus, I feel the Zn reduction here is inconclusive. As for the yellow oil being a plant oil: Here again, IMO this is another possibility. The extraction procedure is a variation on STB with no de-fats. Any oils will therefore end up in the final NPS extraction. You state that you tested the presence of oils through de-fats with heavy petroleum distillates and yielded no appreciable amounts. Did you try this with DCM? From my experience, extractions with only naptha (light petroleum) were almost always oily. I also noticed that it is not an efficient extraction solvent, often requiring > 6 defats or extractions to yield any material. I quit using it early on and looked for alternatives. That being said, this property makes it a great re-crystallization solvent. So, I would hypothesize that it is also a possibility that the oil could also be from the plant. Standard A/B extractions with CHCl3 doing 3 defats and 3 extractions will yield a powdery material if the extraction is handled carefully. And lastly, your paper said (if I read it correctly) that you obtained 95 mg of alkaloids from 1200 g of material. That is a yield of ~.0075% If this is if fact correct, some exploration of the process needs looked into starting with the use of light/heavy petroleum as the nps. This seems very low to me, even for wild strains of P. arundinecea. Some of the very good points gleaned from this: Boiling the material is a good way to deal with the chlorophyll. I need to get a wheat grass juicer =). And that extracting usable material from a plant with a low alkaloid content might be feasible. Quote:The alkaloid exists in the plant as fb? Your notes would seem to indicate this. This was always my understanding based on my early reading about extraction of alkaloids. Thank you for sharing your information and please don't take my critique harshly, it's not meant that way. It is only meant for clarity and understanding. Keep up the work, it gets Phalaris one step closer to being more well understood and a viable source of alkaloids.
|
|
|
DMT-Nexus member
Posts: 465 Joined: 18-Jan-2008 Last visit: 09-Nov-2025
|
Quote:While this would definitely help things to crystallize, it throws another variable into the mix that complicates things. My thinking here is that by performing this step, you will not necessarily know if it was any oils in the material or the oil of the oxide that was preventing crystallization, you've effectively dealt with both at once. Side by side comparisons of the same material would lend more evidence to one or the other possibilities. That is, do one with only the reduction, do one with only the defat, and one that incorporates both. Then compare results. Dozuki, initially I performed re a/b's on the material with defats, no change. I have also run the Zn reduction without an intermediate defat step; half of the freeze precip. samples were produced sans defat, I only added it in after running into 1 or 2 samples which were more oily- possibly a result of stem material getting caught up in the juicing process- where the product precipitated but quickly became oil on reaching room temperature. From the top of my head I remember the sample photographed with the hair grip was purely the result of Zn reduction with no defat performed whatsoever. Quote:95 mg of alkaloids from 1200 g of material This is of course wet weight. Taking that down to dry weight equivelency 95mg from 120g- or there abouts- doesn't seem too bad for a source that can be harvested in 8 weeks or so mistakes were made
|
|
|
Faustian Phytochem Investigator
Posts: 194 Joined: 31-Oct-2011 Last visit: 29-Nov-2025 Location: Oaxaca
|
Quote:Taking that down to dry weight equivelency 95mg from 120g- or there abouts- So then the equivalent dry weight yield would work out to .075%. This, according to the literature on P. arundinecea should work out to .0375% if the material were actually dried as the yield typically drops in half with drying (can provide ref. if wanted). We don't know if this also holds true for P. bracystacys, but best not take chances. Marten et al (1973) found the alkaloid range of % dry material in arundinecea to be between .02 to 1.19% in individual clones grown out in diverse conditions. This puts your grass on the lower end of this scale. P. brachystachys is thought to be higher yeilding with Festi et al claiming an extreme 3%. Despite yields, your work provides good news. I just wander if higher yields and elimination of the difficult oil could be worked out by modifying your procedures. Really, all I'm trying to suggest is that higher yield may be possible 
|
|
|
analytical chemist
Posts: 7463 Joined: 21-May-2008 Last visit: 09-Aug-2025 Location: the lab
|
jamie wrote:benzyme wrote:very nice account indeed.
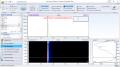
I did a sonication on some phalaris a while back, followed by a quick A/B. the product looked identical after re-x. Brachystachys is certainly the way to go, yield-wise. So did you bioassay it? I did not, but I ran LC-MS on it. Signal intensity was 90 for dmt parent ion and 10 for gramine "Nothing is true, everything is permitted." ~ hassan i sabbah
"Experiments are the only means of attaining knowledge at our disposal. The rest is poetry, imagination." -Max Planck
|
|
|

Kalt und Heiß, Schwarz und Rot, Kürper und Geist, Liebe und Chaos
Posts: 4661 Joined: 02-Jun-2008 Last visit: 19-Apr-2026
|
fourthripley wrote: Dozuki, initially I performed re a/b's on the material with defats, no change. I have also run the Zn reduction without an intermediate defat step; half of the freeze precip. samples were produced sans defat, I only added it in after running into 1 or 2 samples which were more oily- possibly a result of stem material getting caught up in the juicing process- where the product precipitated but quickly became oil on reaching room temperature. From the top of my head I remember the sample photographed with the hair grip was purely the result of Zn reduction with no defat performed whatsoever. Fourthripley, first off, thanks for the experiments and the reporting back, as everyone else has said! I totally love seeing people experimenting - this is what pushes the field forward, you're carving the future! But on the other hand, any claims of "oxide" must be taken with a grain of salt if you do not really make a first hand demonstration of it. I am confident that you know your stuff, but things are often misleading. This is totally OK; I have also stated in the past (and not only once!) that the yellow is oxide, that the oxide is extracted and so forth. As the Nexus is slowly moving away from spot tests and subjective observations, so any claims must be backed up by evidence, not indication. Every knows the frustration of having a million of indications that things are "convincingly" as stated but really, for as long as there are alternative explanations to your experiments you should not refer to the presence of oxide and it supposed reduction with zinc to dmt. Color changes mean nothing. Think about this; let us assume that what you extract is just dmt, devoid of any plant fats whatsoever. Let us also assume that this dmt can be oxidised to create what appears to be a yellow oil; you still do not know what that oil is. You do not know whether it was extracted from the plant as is or whether it formed during the extraction. But let us assume that it is an oxidation product. You still do not know whether it is dmt N-oxide (the stuff you can reduce with zinc) or another oxidation product of dmt that you cannot revert back to dmt (as an extreme example, by literally burning dmt you oxidise it in a manner not reversible with any reducing agent...). Throw in the added complexity of that the reduction reaction per se can discolor stuff (as endlessness said, yellow brand vinegar loses totally its scolor just by adding zinc to it - we did that together and we were both surprised to find out) and you really have to fight a long way to convince anyone that what you have is dmt-n-oxide that you reverted to dmt by zinc without any real proof. Other that these few pointers, keep up the good work! Need to calculate between salts and freebases? Click here! Need to calculate freebase or salt percentage at a given pH? Click here!
|
|
|
analytical chemist
Posts: 7463 Joined: 21-May-2008 Last visit: 09-Aug-2025 Location: the lab
|
yeah, a lot of the n-oxide stuff is speculative LC-MS would give a better picture, especially in the positive mode. if there were an oxyanion bound to the amine, it would likely get protonated and dissociate in the ESI process; the fragment may then be detected in EI mode (assuming the hydroxy formed from protonating the oxyanion doesn't dissociate in acidic solution used to prepare the sample...then it would be impossible to discern fragments from the acidic mobile phase) hmm.. *strokes goatee* I think derivitization would be in order. Was Burnt ever able to show N-Oxide in GC-MS? we need some goddamn H-NMR data "Nothing is true, everything is permitted." ~ hassan i sabbah
"Experiments are the only means of attaining knowledge at our disposal. The rest is poetry, imagination." -Max Planck
|
|
|
DMT-Nexus member
Posts: 14191 Joined: 19-Feb-2008 Last visit: 20-Jun-2026 Location: Jungle
|
Today I put some dmt in a petri dish, added 10% hydrogen peroxide, some of the dmt dissapeared/dissolved (was funny to see some of the dmt crystals moving about fastly through the h2o2, as if they were "boiling" ).. Then some remained undissolved, so I added some IPA and a bit more h2o2 and again the remaining crystals started moving around fast and dissolved. I let it stand for a couple of hours, and evaporated with a hair drier, it left an orangeish oil that smelled sweeter than DMT. I sent this for GC-MS (EI), I'll let you guys know the result if it shows as something different than DMT relevant TLC plate from this experiment posted here: https://www.dmt-nexus.me...m=312045&#post312045
|
|
|
analytical chemist
Posts: 7463 Joined: 21-May-2008 Last visit: 09-Aug-2025 Location: the lab
|
I doubt TLC would differentiate N-Oxide with even halfway decent resolution. this is a job for LC and the expensive voltmeters, to get a more accurate assessment..particularly using IEC (ion exchange chromatography). "Nothing is true, everything is permitted." ~ hassan i sabbah
"Experiments are the only means of attaining knowledge at our disposal. The rest is poetry, imagination." -Max Planck
|