Since ages I was wondering why Bufotenin degrades in very alkaline media and what might happen here. Now I found some time to take a look into that last question that I had. Therefore after now, dont expect too much stuff posting from me anymore.


I cannot find original source, but 69ron was mentioning it somewhere. After this reaction it is not active anymore, so you would destroy your Bufotenin when handling in this environment.
This is also the reason why using at most Ca(OH)2 or Na2CO3 instead of NaOH to freebase the alkaloids from A. colubrina or A. peregrina.
Now obviously it has a (phenolic) Hydroxy group and which should be responsible for this. Also this group would be acidic at very high pH and not at medium values, where the typical alkaloid amines start to react.
So a reaction that might be possible and which I could imagine:
That alcohol only is deprotonated at high pHs and thus could push its electrons inwards, causing some change with that pi system, in case any suitable oxidation agent is present. If it's a fast reaction, then even plain Oxygen could do that and this is basically everywhere.
That molecule looks totally different from a tryptamin and will not have the same pharmacodynamic. So therefore that might render it inactive? Any Keton will be visible in 13C-NMR because of some new signals at really high ppm ranges of 180 ppm +. That would be a super easy way of prooving that reaction.
Now to get any insight I was doing what I wanted to do since ages. But as I dont have any MS/HPLC by hand anymore, I had to wait for a friend to help me out.
Reaction:200 mg Freebase Bufotenin is placed in 5 ml of 10 % NaOH in Water. Indeed the Bufotenin is the only Tryptamin to dissolve here (except Psilocin/-cybin if anyone would have some), as it is ionic above pH 11. After already some minutes the brown mixture becomes very dark. The mixture was stirred for 24 h at RT. Then I did a TLC to check it. New spots, but still mostly Bufotenin. So I stirred for another 24 h and still mostly Bufotenin. Now a last 24 h of stirring at RT. Did not want to heat it up, because wanted to see if even just "storing" it at that conditions will already destroy it. So after a total of 72 h the final TLC also had no different look, so I thought that this is where it might stop.
Here is the TLC after 3 days at RT in 10 % aq NaOH. Solvent was Methanol + 2 % Ammonia.
(All Anions would normally not run at this conditions as they would need to be "freebased but with an acid". I tried therefore using Methanol + 2 % of a pH 10 buffer which would freebase all phenolates. But the DC looked the same, so they seem all to be freebased by the silica powder already and just the standard eluent seems enough.)
You can see 2 different wavelengths, one really high energy (254 nm) where nearly all "medium size" molecules start absorbing and will be detectable. The other one is of lower energy (320 nm) but still responsible for you getting tanned when being in the sun ... just not getting full sun damage anymore. Here only kind of bigger molecules absorb light and also only with suitable electrons. Normal DMT would not for example.
Observation:Always to the left you see plain Bufotenin. It is also still quite the main compound at 254 nm. Besides some smaller spots are seen at the bottom that dont move too much. This is always a good hint for oligomeric tryptamines, they would always be stuck there. Other than that it could be more polar stuff. A ketone (like in the reaction scheme above) would have a higher dipol moment than an alcohol even if people would assume alcohols to be the most polar standard group. But no idea if it would run that low because of that ...
In any case quite some new spots but it looks by eye still like mostly Bufotenin. Sadly I think that when starting with 200 mg I might never get enough material to see that traces in NMR. Therefore I now only relied on some HPLC and GC results.
Now I extracted it with ethyl acetate. Normally any Bufotenate (which should be the regular state of Bufotenin at pH 13) would not migrate. Indeed the Ethyl Acetate only looked moderately brown-transparent, while the aq phase stayed completely black. But now when adding some pH 10 buffer to the water (which should freebase all Bufotenates) the next EA extraction was not stronger colored. I did a TLC of the EA and the water and both looked the same. So I was sure that the EA contained all the relevant compounds and as they are too less for NMR, it would still not be a problem for MS if I only have traces.
Sadly the stuff of my friend is solitary and not coupled, meaning only single-injection for MS and no analysis on that HPLC peaks, but we may still be able to get some good insights.
HPLC:Here is the chromatogram, done with Reversed-Phase meaning eluting on a non-polar column with Water to Acetonitril.

The column was quite overloaded, but then we can also see the traces a little bit better on the other hand. Bufotenin comes at ~ 4,5 min and so there are some peaks before and after. Mean other stuff is at t = 1,47 min; t = 3,04 min; t = 5,47 min; t = 5,87 min; t = 6,00 min. Their ratio is roughly 1 : 0,8 : 0,5 : 0,5 : 0,7. And all of these are only traces compared to Bufotenin.
MS:He told me it was done with ESI-MS. Here are all the negative charged ions:

He told that high-molecular stuff is most likely an old pollution. So just ignore that. Other than this we only see the same Acetonitril-Adduct like for DMT
here. So no special stuff ... No molecule peak for the plain Bufotenate (if that name exists) ...

Here are all the positive charged ions. That is where it's more interesting concerning alkaloids:
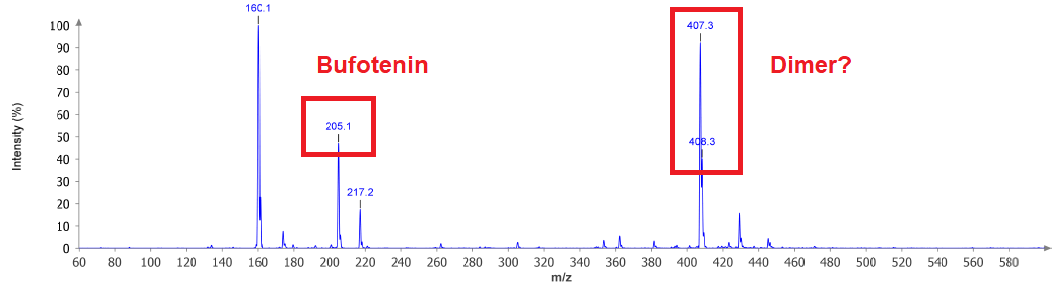
We can see Bufotenin and also what could correspond to a Dimer. Sadly I have not too much idea about MS and therefore dont know if that +12 u has any deeper meaning. I think -45 u could be the cleavage of Et2N from the molecule. That's kind of reasonable. This would mean this peak is also no other compound and just evades from fragmentation. Other than that the interesting thing here is: NOT too much interesting things. This means that instead of having an explosion of components due to the long-lasting and harsh conditions there is not much happening.
But now we can see that possible Dimer. In conjunction with not too much other stuff in MS, but MANY spots at the TLC at very low Rf there is one thing coming in my mind:
What if all of the reaction from Bufotenin is just an Oligomerization?That would explain the following stuff:
- Oligomers will not vaporize and be inactive
- If these Oligomers are more polar then it would reduce the already low bio-activity of Bufotenin even further, even if you WOULD evaporate them
- Oligomers will appear only as multiples in that MS and therefore no wonder that most of them are out of the scope of the diagram above
- If their pi-System is linked, Oligomers will have a much bigger Chromophore. This means more pi-Coupling and therefore absorption of even lower-energy electrons aka red-shift. In addition, different sized Oligomers will have different absorption / fluorescence properties and appear as different colours throughout the TLC.The last one is especially interesting, because we can see some blue fluorescence at 320 nm. This fluorescence is only something which you would see with a big chromophore aka big pi-System and not for regular tryptamines.
So that is why I would now say: There is no phenolic-induced basic degradation. What happens is the simple oligo-/polymerization of Bufotenin that was already pointed out here in the Forum, taking place at basic conditions.
Now further: Why is it not happening for DMT aka not too severe polymerization in pH 13 water?
The simply answer might be: As DMT can not be dissolved in pH 13 water and only stays finely suspended, it will not be incorporated in the media like Bufotenate and therefore not have the same mobility/reactivity if not solvated. Therefore keeping Bufotenin in pH > 11 aqueous media might induce the fastest polymerization of all "regular" Tryptamines. Now why is the degradation when preparing the Bufo Paste from A. colubrina more severe? Probably because when drying the paste the whole mixture is much more concentrated than in this voluminous solution and therefore shifting the reaction towards polymers.
A polymerization reaction would look something like the last picture
here at the bottom of the post, originally from
this source.

With that mechanism, multiple chain length of Bufotenin would be formed, causing
multiple variants and intensities/colors of
Fluorescence at 320 nm and therefore also
multiple heights of TLC spots while the original MS does
not show any explosion of new chemical species. Not sure if this is the absolute truth, but all the results here point to that direction.