
DMT-Nexus member

Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
This is a Compilation of chemical properties of the Nexus' members most common molecules to come across. Not all are Psychedelics, but some are just other imporant molecules or derivates. People searching the internet for temperatures to evaporate DMT or similar compounds will quickly stumble upon values on Wikipedia like a boiling temperature 160 °C at 0,0008 bar for DMT and even 320 °C at 0,00013 bar for Bufotenin. While this temperature physically corresponds to the boiling point like water at 100 °C, you can evaporate those Tryptamines actually way lower than their boiling point, which might make people do wrong assumptions about the vaporization behaviour of those substances. A great explanation for this can be read in depth by another member, check out here! Use Vaporization Temperature given here to exactly vaporize your goodies without combustion at the minimum heat required - in case you have any setup that controls temperature precisely, which is highly recommended for anything. Interestingly, the data even seems to hint that a wide range of tryptamines all evaporate at roughly the same temperature window. Read some philosophy behind that idea here. Use Solubility data for adjusting solvent for recrystalization to not overshoot and get less product back. Use Vaporization data to set up your vaporizer correctly to get the biggest hit while not burning your material. Some molecules have bonus data / discussion for various reasons. There is already a page on the wiki, but this data is 99 % new and so I would also like to post it here, as it may be more accessible for people who rarely check the wiki. Now also included an UV/Vis spectrum, measured in Acetonitrile. Now also included an FTIR spectrum, measured in ATR-mode. Now also TGA / DSC added as a more scientific take on phase transition analysis than hotplate experiments. Quick note on TGA: You can fully evaporate a substance even at the start of the TGA evaporation curve. Evaporation is a function of surface area and ventilation. Therefore even the onset temperature of a TGA can be used to fully evaporate DMT at 160 °C for example. The end of the curve simply implies if the substance can be evaporated completely or will also partially decompose. Current content:
|
|
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
DMT  Solubility:boiling Naphtha (80 °C, C6-C7, 60-80 °C) " infinite" (the DMT will melt and just create a homogeneous liquid-liquid solution. In the end it may even be a solution of Naphtha in DMT) Example: In just 5 ml of boiling Naphtha you could easily add 4 g of DMT, while only doubling the initial volume. No precipitation would ever show off. Naphtha (20 °C, C6-C7, 60-80 °C) 29,3 g in 1 L 2,93 g in 100 ml Naphtha in the freezer (-20 °C, C6-C7, 60-80 °C) 1,09 g in 1 L 109 mg in 100 ml boiling n-Hexane (69 °C) " infinite" (same as above for Naphtha) n-Hexane (20 °C) 7,7 g in 1 L 770 mg in 100 ml n-Hexane in the freezer (-20 °C) 825 mg in 1 L 83 mg in 100 ml Here 2 additional Hydrocarbons: n-Pentane in the freezer (-20 °C) 501 mg in 1 L 50 mg in 100 ml n-Heptane in the freezer (-20 °C) 916 mg in 1 L 92 mg in 100 ml n-Heptane can be heated to 98 °C and therefore above DMTs melting point. That's why it can also potentially hold infinite DMT at boiling temp. For n-Pentan it's not possible (already boiling at 36 °C so the maximum will be quite low. For recrystallization just go with whatever Hexane/Heptane/Naphtha is the cheapest to get. But always make sure to get a Naphtha that is a mix from Hexan/Heptan isomers. Besides very soluble in Room Temperature Acetone, IPA, Ethanol, Methanol, DCM, Et2O, MEK, Toluene, Xylene, DMF. Phase Transitions:There are at least 3 reported crystal structure modifications of DMT, coming in different crystal grids and thus different melting points. I just know the one that precipitates already at room temperature more yellow and the one that precipitates at lower temperatures fully white. Hence I believe the later one might be more crystaline as it takes longer and slower precipitation, so the melting point might also be higher. Measured with IR Thermometer on a hot plate (give it +- 5 %) Melting Point 58 °C Melting Point - DSC (see further down) 63 °C First fumes starting 100 °C Strong fumes from 160 °C+ No fumes arising anymore 190 °C I think 175 °C is optimal for vaporization.IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (0,05 mg/ml in Acetonitril) 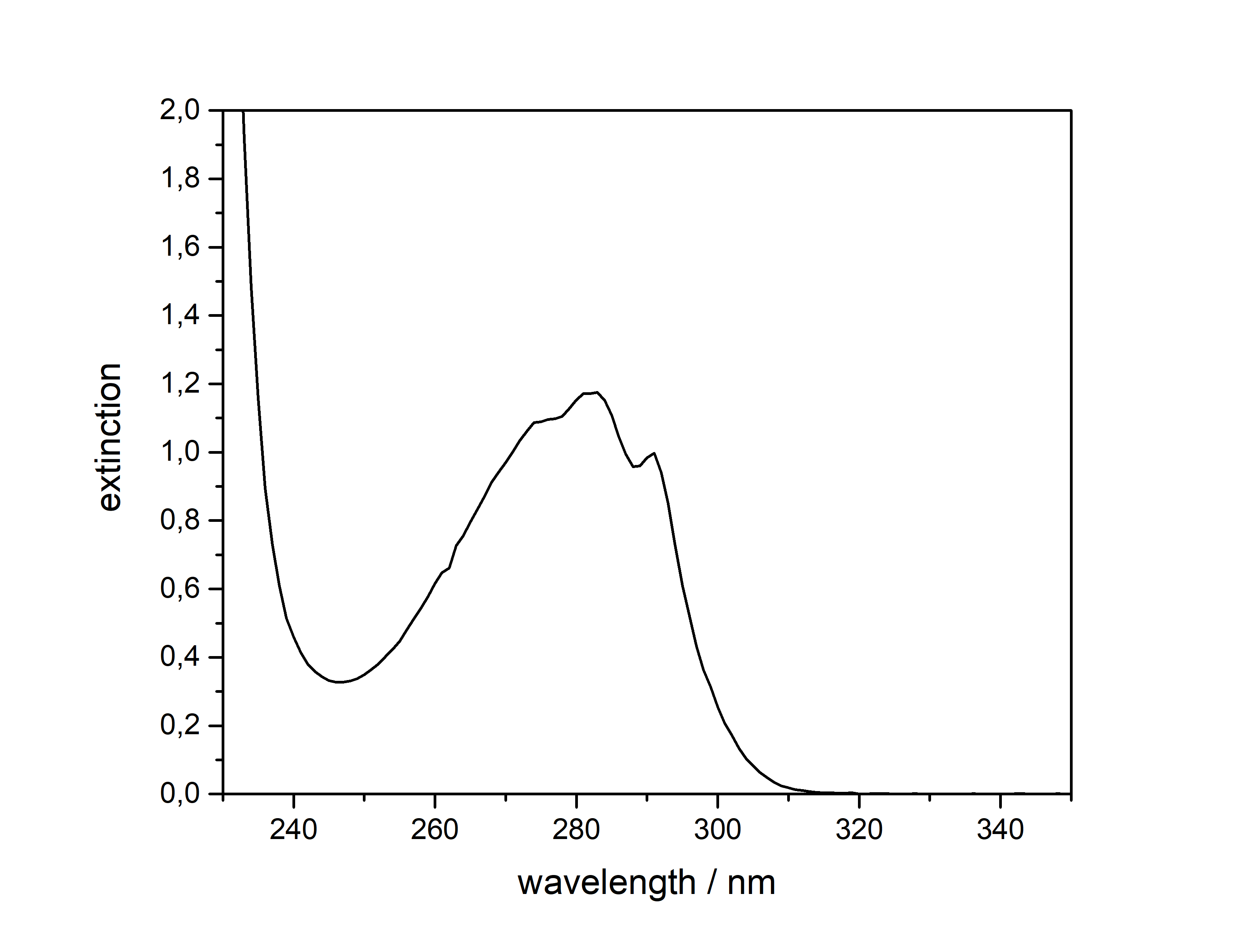 No absorption in visible range (400+ nm), so it is white ... But what you can see here nicely is that it just got that typical tryptophane peak at 280 nm - this peak is normally used for protein UV-detection. Thermogravimetric Analysis (TGA) (Heating rate = 3 K / min) Here is the thermogravimetric analysis of DMT. Heating rate is 3 K / min. What you can see here is the mass loss of a sample while heating. Thats very nicely demonstrating any evaporation effects. So what you see here: First Vaporization starts at 145 °C (even though I can see trace vapors coming from DMT at 100 °C) and ends at 232 °C. I guess the formerly proposed vaporization range of 165 - 180 °C still seems to be the best choice. Differential Scanning Calorimetry (DSC) (Heating rate = 3 K / min) Here is the differential scanning calorimetry analysis of DMT. Heating rate is 3 K / min. What you can see here is the uptake or release of Enthalpy of the sample while heating. Thats very nicely demonstrating any phase transition effects like crystalization / melting. A rising graph indicates an endothermic process, which consumes energy and a decreasing graph vice-versa. So what you see here: There is a melting point onset of DMT at 63 °C. No further melting points detected, so we dont have any polymorphism in this sample. There is another endothermic effect between 200 °C and 245 °C. No idea what that is, but maybe some sort of thermal degradation? Still I think that should only happen > 250 °C ... anyways as the TGA suggest vaporizing your DMT at < 200 °C, this once again shows that for efficient / healthy / non-material-wasting evaporatoin the stuff should not be heated > 200 °C. Comments: Not much to say. Dont heat your Spice above 200 °C, this will just cause combustion at some point. Therefore approaches with open flame and Changa will always create unhealthy smoke, that bad charred flavor will just be hidden by the strong DMT-smell. Better use a controlled way of delivering 160 - 200 °C and inhale fresh healthy swiss-alps-DMT. Also that solubility in the freezer value may now help you to plan your re-x. No more " Maybe you just have to evaporate more Naphtha to get any product?  " Just estimate that ~ 1 mg of Spice will be still held back per 1 ml of Naphtha after freeze-precipitating. This means dont re-x your 100 mg Spice in 100 mg Naphtha  Also if you haven't got any precipitation from 100 ml of Naphtha in the freezer, then you still wont get more than 100 mg from that even if you further reduce ... maybe the problem happened earlier then.  That UV/Vis-spectrum is not that interesting, but it shows exactly the Tryptophane-Absorption at 280 nm, which is also used for Protein UV-Detection. Nothing special else though. If more polymorphs may be accessed with different melting points, these could be measured by XRD and then maybe higher-melting-point-spice will also show greater degrees of crystalinity. It would be senseful if very amorph Spice would be the reason for people observing DMT melting as low as 50 °C and high-crystalinity Spice would be causing 80 °C melting reports. Nevertheless that 58 °C sample looked also quite crystalline ... Active Close-Up: (that left thing looks quite like a micro eagle) For more deep-look images check either here, or to basically see the DNA of DMT by electron microscopy check this.
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
DMT-N-Oxide  Synthesis:Dispense 1 g of DMT in 2,4 ml of 30 % H2O2 (does not dissolve as this is basically water). Add amounts of Ethanol while stirring until all DMT is dissolved. Should be ~ 7 ml Ethanol in total. Stirr this mixture for 48 h at Room Temperature until full conversion. Evaporate all solvent, leaving behind DMT- N-Oxide. ( source) TLC after 48 h still showed a spot in height of starting material. Nevertheless the final product was extracted with boiling Hexane and this Hexane showed no clouds when Trifluoroacetic acid was dropped inside, indicating no residual Freebase so conversion was quantitative. Also the majority of solvent was evaporated by N2-stream. The last bit was evaporated on a hot plate ( Picture 1). This should be done with caution, as it was discovered the N-Oxide is very heat sensitive and decomposes already above 110 °C ( Picture 2, turning completely black just within less than 3 seconds). If treaten with care it will form a waxy orange solid ( Picture 3). Solubility:boiling n-Hexane (69 °C) insoluble Aceton (RT) insoluble DMF (RT) insoluble DMSO (RT) soluble Water (RT) soluble Wow quite bad solubility overall ... Still its soluble in even neutral water, given the properties of being a salt and as usual in DMSO, which is the best overall solvent anyways. Phase Transitions:Interestingly pure DMT- N-Oxide is an orange solid and not an oil contrary to popular believe. The oily consistence only arises due to always encounter it in regular ways in a mixture with DMT. Already small mixtures of substances make them not only prevent to crystalize, but also often forming an oil - this sample for example is an oil, yet 80 % DMT purity - even though they may both be an actual solid when being pure. Measured with IR Thermometer on a hot plate (give it +- 5 %) Melting Point 40-50 °C hard to determine due to waxy structure DMT- N-Oxide turning black 110 °C First fumes starting 130 °C No fumes arising anymore, black tar residue 150 °C I think there is no optimal temperature for vaporization. It just decomposes super fast, even on inert surfaces like glass. It does not even give strong fumes, it mostly turns into black tar. That is why even if you HAD some N-Oxide in your mixtures, it would never reach your lungs probably. IR-Spectrum (measured in attenuated total reflection method (ATR)) DMT-N-Oxide separation:As the N-Oxide seems to be only soluble in DMSO, this can be achieved to do a purification from other possible tryptamine compounds. Yet it seems this can turn oily stuff into more solid products, but yet I would not call this an actuall recrystallization.
1. Dissolve the N-Oxide in a minimal amount of DMSO
2. Add 20x Volumes of Ethyl Acetate (possibly Aceton could work, too)
3. The solution turns cloudy, place in the fridge for 3 h
4. Decant the clear liquid to retrieve your N-Oxide (Picture below). All non-oxidized Amines will stay in solution, no matter what Tryptamines they may be. Comments: So interestingly DMT- N-Oxide is a solid, but thats also what you can observe with DMT: Just having small traces of impurities it will already start to become oily. Still, the substance is quite waxy. Also interesting, but annoying seems the super fast thermal decomposition, starting from 110 °C and running super fast, finishing in seconds. If just started once you will lose your N-Oxide 100 %. This makes it super unlikely that anyone ever has smoked some real N-Oxide, also it seemingly being non-soluble in Acetone as it seems, preventing it to be transfered further into Changa. People still dissolving all their oily DMT in Acetone probably had no N-Oxide pollution, but just oily DMT due to other impurities. Actually I even think having any reasonable of the N-Oxide in your extracted stuff is just super unlikely. Why should it have become oxidized? You would need a real oxidation agent or UV-rays and pure heat does not oxidize DMT - even when in direct contact with O2. Also UV-Rays will not make a full conversion. Therefore even if there were traces of N-Oxide in a plant-derived sample, they will always come with a big bunch of non-oxidited DMT. Only a real chemical conversion would lead to 100 % N-Oxide. So based on smoking oily residues labelled as N-Oxide and being called active, this may have just been a false positive thing. I more have the feeling that people in general call oily DMT being polluted by N-Oxide, but also even small amounts of pollutants will turn your goodies to an oil, this sample is a classic oily mess and still 80 % of DMT Purity. In regard to bioactivity of DMT- N-Oxide I think its nearly impossible to smoke this compound, as it will turn black within less than 3 seconds once heated above 100 °C. By that time it will not even have evaded any fumes to inhale. Further heating does just produce traces rising up from that super uggly and unhealthy looking black tar (see Picture 2). But as the N-Oxide is the natural product of MAO-enzymes, it should not be needed to block these when incorporating the N-Oxide. This means if it HAD any psychedelic effect one could simply eat any amount of N-Oxide and would suspect an effect not too far off from the same amount of DMT, eaten with 150+ mg Harmalas. Now people could argument, eating would be an inefficient ROA, as the high-polarity compound is not likely to tresspass membranes and will have low bioavailability. Actually it is even less polar than DMT when eaten orally: macroscopically the positive charge is decreased due to the additional Oxygen at the protonated N-Oxide. And now DMT is indeed bioavailable when eaten, as people know so the N-Oxide should be too, if it had any activity at all. The proof can be seen here in this reversed-phase Chromatogram. The solvent protonates any bases, causing them to have uniform charging across the column. This happens to DMT and the N-Oxide, while now being DMT+H(+) more polar than DMT-N-OH(+). In a consequence, the N-Oxide elutes 1 min later in reversed-phase and the solvent can be transferred to our gastric conditions, where also both compounds will form their protonated salts. This way I am very certain, that if N-Oxide HAD an effect it will be visible upon oral ingestion. 80 mg of DMT- N-Oxide was eaten. No effect  But: It tastes like DMT (Ginger-like spicy) + an additional Cherry Flavor. Super cool! Weird sweet bonus, exactly like Cherry Menthol gums. So while the N-Oxide being a metabolist of DMT and definetly has a metabolic pathway in the body, I highly doubt it would still interact with the same receptors that are also responsible for DMT-effects. That unoxidized Amine would normally be important for in-vivo recognition at receptors, rendering it possibly inactive upon oxidation, as the N-Oxide is a very different chemical species - unlikely to show the same binding behaviour. That said and given the facts that it cant really evaporated and shows no effect upon ingestion, I highly doubt that anybody ever was high on N-Oxide .
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
DMT-Acetate  Solubility:Very soluble in water. Insoluble in all organic solvents - except when used in excess (IPA, Ethanol, Acetontril and comparable). DMT Benzoate is soluble in DMSO and DMF - so DMT Acetate probably too. Phase Transitions:Many people have wondered and debated about whether DMT-Acetate can be smoked direclty. Common sense would tell it is a salt and would take insane heat to vaporize. Also it would be caustic to the throat and lung as it is still 1/4 Acetic Acid. So here there is some actual data about this: Measured with IR Thermometer on a hot plate (give it +- 5 %) Melting Point / First fumes starting 130 °C Strong fumes from 165 °C+ No fumes arising anymore 190 °C I think 175 °C is optimal for vaporization.IR-Spectrum (measured in attenuated total reflection method (ATR)) Water-Uptake It was reported only -Fumarate/-Maleate/-Benzoate salts are the common variants that are not hygroscopic (maybe Ascorbate too?). I'm not sure whether DMT-Acetate really is also hygroscopic, it may just already be an oil in general - it seems to me - even though thats anti-intuitive, being a salt itself.
Still this could be verified very easily by NMR. Water appears in 1H-NMR at 4,67 ppm. This means you can determine the water content very precisely by calculating: Integral 4,67 ppm (s) = I(w) Integral 2,27 ppm (s) (CH3-) = I(D) 1. Calculate Molecule ratio of Water Molecules to D-Acetate-Molecules: = (0,5*I(w)) / (0,16*I(D)) 2. Calculate Mass Fraction of Water Molecules to D-Acetate-Molecules: 100 * (18/248 ) * Molecule ratio = water content [%] Example: if 1H-NMR shows an Integral of 2,75 at 4,67 ppm (s), then the water content in DMT-Acetate is 10 %. But well thats just a random example and not measured. I would have access for measurement, but I dont feel quite well to do that right now ...
In theory water could also at least qualitatively identified in that IR-spectrum. There should be a big peak at ~ 1600 cm^-1, but that could also be from acetic acid. So not really sure, but I also dont really have a Clue about IR. UPDATE: Now did that test and also find it here. Same test for many more salts found here. Comments: So it seems that DMT-Acetate can indeed be smoked, just based on having the same vaporization range as Freebase Tryptamines. Quite interesting that there is practically no difference to DMT. It both starts to emit strong vapors above 150 °C and stop vaporizing below 200 °C. Therefore it does not seem to be retarded strongly by the fact that its a salt. Maybe it may be explainable by counting Acetate as a soft ion and not a hard ion like chloride or sulfate. Bioassay: A small portion of DMT-Acetate was vaporized according to the optimal vaporization temperature. The vapor was not caustic and no acetic acid smell was present, neither it was unpleasant to the lungs. A regular DMT-like effect was felt. If it had the same potency, hard to say as this oil is hard to weigh. It would be possible to weigh a loaded apparatus and weigh it after the dose was taken. As normally even the tiniest amounts of Acetic Acid are very irritating to the lungs, it seems there is no back dissociation form Acetate to Acetic Acid and the Acetate is not irritating to the lungs. This also means that the DMT will be delivered to the Lungs as a salt and even this protonated form shows some promising metabolism. This is remarkable as other compounds are often blocked for desired physiological pathways when not freebased. Still, being a slushy oil, this is not a convenient form to handle your spice. PS: It seems hard ions like the sulfate are indeed not vaporizing and just baking to a black cake, no fume release even after 250 °C. So it may be that the Acetate is just being able to be smoked because of being regarded as a soft ion. So next I checked the Benzoate salt version to also verify on this one. DMT-Benzoate  Solubility:Very soluble in water, DMSO, DMF. Caution: In contrast to DMT Fumarate it also has a high solubility in organic solvents. In general already mediocre at room temperature, but excess Ethanol / IPA will quickly dissolve it. It can be even dissolved in boiling Aceton / Ethyl Acetate. Therefore take care when playing around with this salt - you might actually loose some otherwise. Therefore you would need a much higher concentration of DMT / Benzoic Acid than when creating the Fumarate-salt instead. Much better than the previously mentioned solvents is Toluene, which also requires freezing to get most DMT Benzoate out as a precipitate. According to Loveall also Limonene has a really low solubility at room temperature so Limonene is probably the best solvent to create DMT Benzoate, as no freezing is required and the solvent is not toxic like Toluene. Still for a purification by recrystallization Toluene might still be better, but solubility of DMT Benzoate in boiling Limonene was not checked and maybe is also high enough. boiling Toluene (110 °C) 4 g in 100 ml 40 g in 1 L Toluene in the Freezer (-20 °C) 250 mg in 100 ml 2,5 g in 1 L boiling Hexan (69 °C) 1,4 g in 100 ml 14 g in 1 L. Hexan in the Freezer (-20 °C) < 200 mg / 100 ml < 2 g / L Also Hailstorm found that Amyl Acetate works great: Hailstorm wrote:Another way to purify DMT benzoate is to recrystalize it from amyl acetate (which is also a non-toxic non-polar freebase extraction solvent with a logP very close to that of DMT) taking care not to heat it much above 100 °C To precipitate DMT-Benzoate, use freezer-cold Toluene or Limonene. Better not use Naphtha for DMT only, as it risks Benzoic Acid precipitating instantly before forming the salt. When using Ethyl Acetate the solution should be concentrated to 1 g / ml first, as otherwise solubility is already too high. Phase Transitions:Same thing as above with the Acetate. Common sense would tell that salts cannot be smoked / vaped. Even more for a more classic salt like the solid, plain white Benzoate. Same thing even when vaporized, it should also give off Benzoic Acid vapors, possibly caustic to the lungs. So here there is some actual data about this: Melting point measured with DSC (Heating rate = 3 K / min). Picture is not included to safe space. Check DSCMelting point peak onset = 163 °C Melting point peak max = 166 °C Based on TGA-DSC-comparison there is not much of a liquid range. The salt directly evaporates after melting. Probably because of the now increased mobility of ions the process is enabled, that let's those molecules vaporize. Thermogravimetric Analysis (TGA) (Heating rate = 3 K / min) Check TGA of only DMT-Benzoate 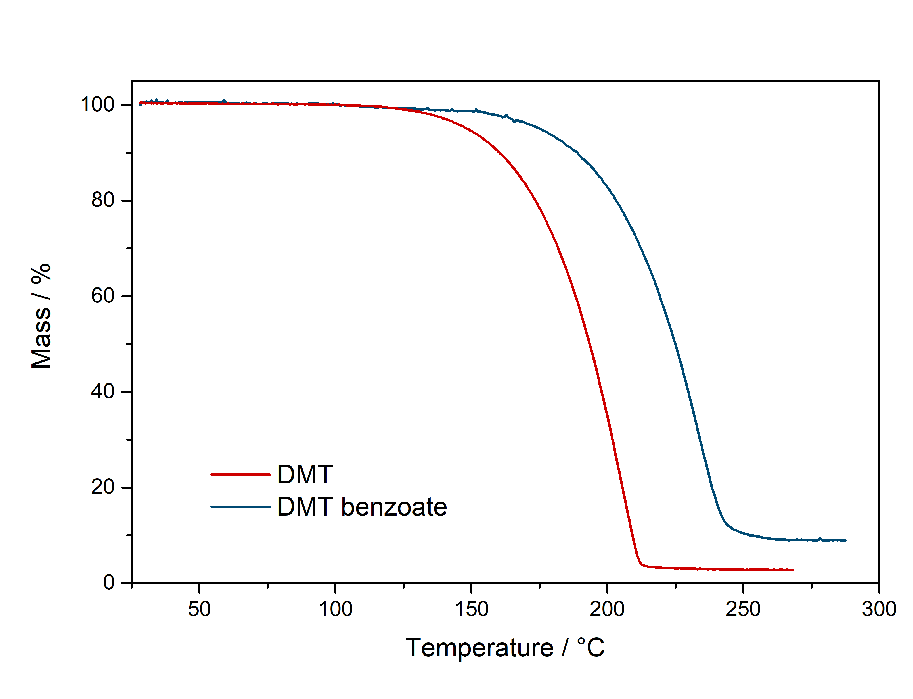 So as expected the melting point for the Benzoate salt is really high, being 166 °C and therefore at least 100 °C above any typical freebase DMT polymorph. That comes quite expected, so I would have also assumed this stuff is never smokable. But then as seen in the TGA, it indeed evaporates really easy. Not only the whole process is simply shifted by ~ 30 °C compared to the freebase, it also evaporates nearly completely. That indicates indeed salts like Acetate and Benzoate are completely smokable, as their evaporation temperature is easily hit with any regular vaping device. Even more if you just use a flame to light it, then you would anyways horribly overshoot as you also do with Change in the Bonga (better not do it, waste of material and ultimately simultaneously burning stuff = cancer). But that means DMT Benzoate would be perfectly smokable if you just exchange your Freebase with this one. Because of this further down I had to test if the vapor also is not harsh - like the Acetate - which I assumed to contain some Acetic Acid Vapour but it didn't. IR-Spectrum + UV-Vis was measured, but not interesting so not uploaded Comments: To me now a question is: Why are these DMT salts evaporating so easily. At least Acetate and Benzoate show nearly no difference to the Freebase. Of course the +30 °C is not small, but it does not matter for practical efforts and I would have assumed like +200 °C.
So far the canon was Salts can't be smoked and indeed the Sulfate is just burned to death. But any practical efforts that have been done on this would have probably been with the Fumarate. I dont have any to test. So it would be an interesting thing to test the fumarate - in case it would indeed not vaporize and be the reason for initial salt-non-evaporation-beliefs then probably this might be linked to the whole ionic complex consisting of DMT*1/2Fumarate instead of being a simple salt like DMT+ Benzoate-. Bioassay: 5 mg of DMT Benzoate were evaporated at ~ 180 °C. Indeed the vapor was not harsh. It gave absolutely no irritation that would come from inhaling acidic components. Only the typical irritation that I get from the freebase ... but then again I'm a noob at smoking. Therefore I would say: You can just smoke DMT Benzoate as you would do with the Freebase. That could now even be employed by people who are super paranoid about the shelf life. Just create the Benzoate and go on in any matter with this compound as you would with DMT - just creating Change would be a pain obviously. More DMT-Salts and also a general perspective on all of these is found here.
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Bufotenin / 5-OH-DMT 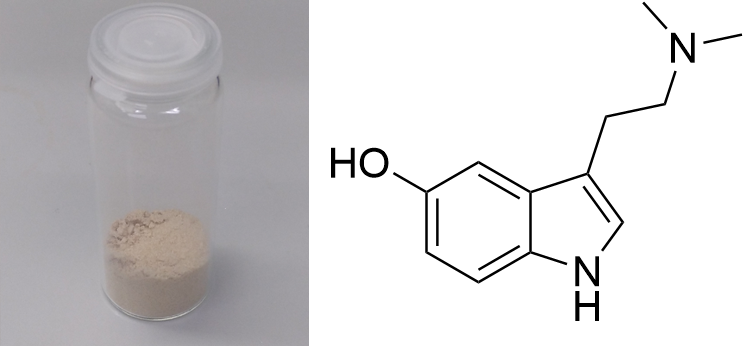 Solubility:Bufotenin is the most polar common Tryptamine, only surpassed by Psilocybin. Therefore it is completely insoluble in Hexane/Naphtha, but can be soluble in pH > 12 water due to Phenol deprotonation. It is hard to crystalize and will mostly give just an oil. The following solutions can give crystalline products: boiling Xylene (140 °C) 43 g/l = 4,3 g in 100 ml Xylene in Freezer (-20 °C) - 20 °C = 0,1 g/l = 10 mg in 100 ml Selectively dissolves Bufotenin and not the more polar components of A. colubrina seeds. But you will need a defat with Naphtha before. With 2x re-x you should get pretty pure and light-colored Bufotenin ( Pictoral). Nevertheless you might evaporate a little more Xylene before placing in the freezer, to retrieve more of your goodies. Why is Xylene a good solvent, even though its way more unpolar than Bufotenin? 69ron gives explanation (cant find source): Xylene can heat Bufotenin above its melting point, thus forming a liquid-liquid mixture, which is miscible. This can also be used with Limonene, but Limonene is much worse as it needs to be heaten additional 30 °C to 175 °C, which causes partial degradation of Bufotenin. 1:3 Ethyl Acetate:Naphtha (C6-C7) Boiling = 7,6 g/l = 760 mg in 100 ml 20 °C = 3,6 g/1 l = 360 mg in 100 ml - 20 °C = 3,5 g/1 l = 350 mg in 100 ml Not as good as Xylene - you should only use it as an alternative when not being able to get Xylene. Still possible to get crystalline product ( Picture). This will still catch up to 2 % of more polar components from the seeds as you can see here in NMR and GC. Also when pulling with this on Anadenanthera colubrina seeds, defat first with Naphtha. Ethyl Acetate Boiling = 280 g/1 l = 28 g in 100 ml 20 °C = 72 g/1 l = 7,2 g in 100 ml - 20 °C = 50 g/1 l = 5 g in 100 ml Mentioned for recrystalization in literature. Not recommended, seems to cause a darkening ( Picture, right). Also very high solubility overall, means you have to use very minimal amounts of solvent = inconvenient to handle. Furthermore if the Bufotenin has a low purity it will just stay oily after re-x. Besides very soluble in Room Temperature Acetone, IPA, Ethanol, Methanol, DCM, Et2O, MEK, DMF Phase Transitions:Measured with IR Thermometer on a hot plate (give it +- 5 %) Even though 146 °C is given in literature, both amorph and crystaline Bufotenin gave the following melting point. Melting Point - DSC (see further down) 132 °C First fumes starting 160 °C Bufotenin turning brown (possible phenol -> keton oxidation) 170 °C Strong fumes from 190 °C+ No fumes arising anymore ... 230 °C ... leaving behind a considerable amount of charred Bufotenin. Seems that it cant be fully evaporated without causing at least partial degradation. I think 220 °C is optimal for vaporization.Interestingly the melting point is really low when measured on a hotplate. Literature value is around 145 °C, but every batch that I checked was well below that. Values ranged from 106 - 132 °C and all being pure by recrystallization. Polymorphism may also be a factor here. When vaporized Bufotenin will inevitably form some black tar which will not vaporize nor dissolve (except in DMSO) anymore. Also the vapor is quite harsh, indicating pyrolysis products being inhaled too. Some thoughts on this and possible solutions are found here. (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (0,25 mg/ml in Acetonitril) In contrast to DMT we can see an elongated absorption further than the 280 nm Tryptophane-peak. Bufotenin has that additional 5-OH, so the hydroxyl group donates some electrons into the Indol-chromophor and therefore changes the minimum required energy for absorption (= electron excitation). Thats why the substituent causes a shift in absorption properties or more precisely an elongation. Thermogravimetric Analysis (TGA) (Heating rate = 3 K / min) Here is the thermogravimetric analysis of Bufotenin. Heating rate is 3 K / min. What you can see here is the mass loss of a sample while heating. Thats very nicely demonstrating any evaporation effects. So what you see here: First Vaporization starts at 182 °C and ends at ~ 275 °C. This is pretty close to what I saw with my Ghetto-Hotplate-Experiments. Sadly it also confirms that instead of fully vaporizing, the Bufotenin will also form pyrolysis products and combust into black tar, which makes any evaporation stop at 50 %. The heating rate is very gentle and only 9 mg of sample were used, so even in that very controlled heating conditions this decomposition cant be avoided. Thats why it will always be a mess when vaporized in any way. Read at the Comments section what I believe is this decomposition. Still it seems the best vaporization range is then 200 - 250 °C. Differential Scanning Calorimetry (DSC) (Heating rate = 3 K / min) Here is the differential scanning calorimetry analysis of DMT. Heating rate is 3 K / min. What you can see here is the uptake or release of Enthalpy of the sample while heating. Thats very nicely demonstrating any phase transition effects like crystalization / melting. A rising graph indicates an endothermic process, which consumes energy and a decreasing graph vice-versa. So what you see here: There is a melting point onset of Bufotenin at 132 °C. Previously with another batch a melting point of 103 °C was measured for amorph and crystaline Bufotenin. But as there were traces of Ethyl Acetate left, this new value was put into the properties table. Literature tells 148 °C if I recall correctly, but this sample here was definetly also free of solvent. Comments: So Bufotenin is hard to crystalize, but easy when using the 2 mentioned solvents. When not heating to 170 °C it seems quite stable and that phenol is not prone to oxidation as people may think. It stays clear white for months even at room temperature. Sadly it does not vaporize cleanly and will degrade by 50 % instead into a black tar. This 5-OH group seems to cause a lot of trouble here not only for crystalization. According to many sources Psilocin (4-OH-DMT) can degrade over the time in mushrooms, as enzymes may partially still work or oxygen also promotes this reaction. It is now shown that this is some kind of polymerization of the indol - read here or here. Although there are no enzymes to convert Bufotenin (5-OH-DMT) it could also polymerize while exhibiting the same Chinon-like structures as Psilocin - only with the restriction to 1 of the 2 possible conjugations, which is an ortho-coupling (instead of para, which is blocked for Bufotenin). Therefore my theory is at > 170 °C Bufotenin turns brown, as it starts - mediated by oxigen - also to polymerize. This reaction creates a pi-conjugated molecule of big size, therefore it will absorb more light and ultimately become black. This also does not vaporize anymore and there you have the residual 50 % Bufotenin as a black tar in your vaporization vessel. Luckily only happens with Bufotenin, as for any other O-substituted Tryptamin (like 5-MeO) the electron density increasing effect is way lower, preventing a reaction like this. The vapor in low concentrations tastes nicely like toasted nuts. In high concentrations it may feel nauseating. But maybe thats just as the overall effect of Bufotenin is a little bit nauseating. Seems this cant be avoided, as Bufo is quite similar to Serotonin and therefore causing intense vasoconstriction and tingling in the face and fingers. Not too pleasant. Also face turns quite cold. Red coloring at the face could not be observed. Still other people have reported very promising / intense / interesting experiences when vaped. Oral consumption seems rather ineffective. 100 mg of Bufo-Fumarate just gave a slight effect, comparable to 1 g of Mushrooms possibly, while still being bad to the stomach. You will need to create Bufotenin Benzoate to smoke it. This will not undergo this combustion reaction and not form harsh vapor. Vasoconstriction is still present to a small degree. See further information here. Close-Up: (it has a round geometry in contrast to DMT / 5-MeO-DMT  )
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
5-MeO-DMT 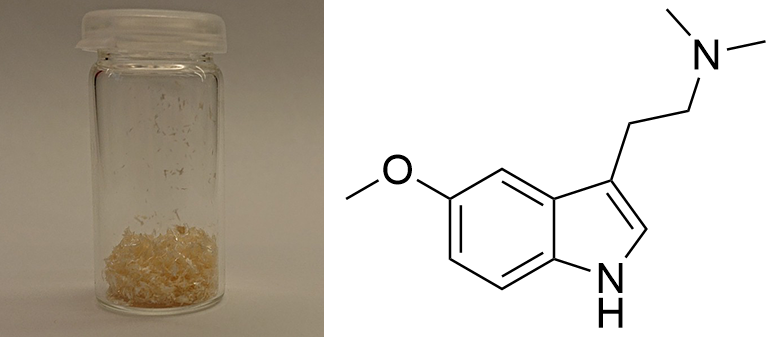 Solubility:boiling Naphtha (80 °C, C6-C7, 60-80 °C)
4,5 g in 100 ml
45,0 g in 1 l
Naphtha (20 °C, C6-C7, 60-80 °C)
104 mg in 100 ml
1,0 g in 1 l
Naphtha in the freezer (-20 °C, C6-C7, 60-80 °C)
70 mg in 100 ml
700 mg in 1 l
boiling n-Hexane (69 °C)
1,9 g in 100 ml
19,0 g in 1 l
n-Hexane (20 °C)
84 mg in 100 ml
840 mg in 1 l
n-Hexane in the freezer (-20 °C)
46 mg in 100 ml
460 mg in 1 l Compared to 5-AcO-DMT the solubility in Hexan is still more than 4x as high, which still dissolves slightly in Hexan. But upon freezing the Solubility drops basically to 0, in contrast to other Tryptamines and their respective solvent-of-choice. So when doing a recrystalization there is basically no chance to loose material, even when only cooling to RT. Also in the freezer you dont get any more precipitation, so just stay with RT. Regarding crystalization, you can find some thoughts here. Besides very soluble in Room Temperature Acetone, IPA, Ethanol, Methanol, DCM, Et2O, MEK, Toluene, Xylene, DMF. Phase Transitions:Measured with IR Thermometer on a hot plate (give it +- 5 %) Melting Point - amorph 47 °C Melting Point - crystaline 49 °C Melting Point - DSC (see further down) 62 °C First fumes starting 118 °C Strong fumes from 170 °C+ No fumes arising anymore 190 °C I think 175 °C is optimal for vaporization.These values are quite unexpectedly low. Regarding melting point, my first idea was that it might be due to some polymorphs and I got a low-melting variant, as literature value is 69 °C. Same was also observed with Bufotenin, where the pure compound showed a much lower melting point on hotplates and in DSC. Nevertheless the DSC of the same Sample is much better in accordance with that value - like Bufotenin. Maybe the Hotplate Test still contained some solvent.
The boiling point is also remarkably low. By adding the 5-OH from DMT to Bufotenin the vaporization temp should have been well above 250 °C, but in this case it was also much lower. But now comparing 5-MeO-DMT to DMT we have nearly the same vaporization profile. This can be even better seen at the TGA measurement later on if you compare both. Also 5-MeO-DMT leaves behind nearly no brown residue, just like DMT and 5-AcO-DMT, indicating that only Bufotenin is a Tryptamin with a *dirty evaporation* which leads to charr. Seems like this OH- of Bufotenin really is the root of all evil, as it can oxidize. IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (Acetonitril, didnt write down concentration) The UV-Vis looks quite like 5-OH-DMT / Bufotenin. That of course comes expected, both share the same electronic properties regarding their chromophore: Oxygen induces +M effect in the Ring, enriching it with electrons. In both cases we see a red-shift of absoption compared to DMT. Still this is not visible for 5-AcO-DMT, here the moiety is basicallys locked from any interaction with that Indol and therefore causes no red-shift.
Still for both MeO-DMT and Bufotenin the absorption has a cut-off before any visible range, so a pure compound will still resemble clear-white. Thermogravimetric Analysis (TGA) (Heating rate = 3 K / min) Here we can see the same observation like during these hot-plate experiments: 5-MeO-DMT just vaporizes like DMT basically, maybe needs only a few K more. Also I was happy to see that it has a really clean vaporization, regarding more than 90 % vaporized at 245 °C. If you compare: 95 % of DMT has vaporized at 230 °C. That is remarkably similar, given that we add this 5-OMe. Furthermore I tried vaporizing a dirty crude mixture. This was still containing most 5-MeO-DMT. But it did only give slight fumes and was moreover burned as a black tar into the hot-plate. So given that your substance has (reactive) impurities the vaporization might be much less clean. Differential Scanning Calorimetry (DSC) (Heating rate = 3 K / min) In contrast to a hot-plate 49 °C melting point there is now a melting observed starting at 62 °C. Later on at 240 °C+ there is another endotherm process, maybe that's also the reason why the last 10 % are not vaporized and instead probably decomposed. Same was observed with DMT before. Comments: So based on these results 5-MeO-DMT and DMT seem like quite close brothers, whereas Bufotenin is more of an oddity.
Regarding phase transition I would have expected at least 50 °C more for vaporization, but we saw that even Harmalas can vaporize at 200 °C.
Seems like an electronic device to be used for the whole spectrum of the Nexians' favorite molecules doesnt even need more than 250 °C max. Temperature. Bioassay:Might try some sublingual stuff in future. Here you can find some dosages for Psychoactivity Threashold derived by Jonathan Ott, quite the same study like above the link at Bufotenin from him. Close-Up: ( Rainbow light refraction when illuminated from top and right-angles  )
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
5-AcO-DMT Seems like I tested an over-acetylated compound here. See more over at the Thread here. There is practically no information about 5-AcO-DMT on the Internet (not to be confused with 4-AcO-DMT). Here is some maybe helpful stuff. Solubility:
boiling n-Hexane (69 °C)
5,0 g in 1 L
500 mg in 100 ml
n-Hexane in the freezer (-20 °C)
1,3 g in 1 L
130 mg in 100 ml
Besides very soluble in Room Temperature Acetone, IPA, Ethanol, Methanol, DCM, Et2O, MEK, Toluene, Xylene, DMF. Phase Transitions:
Measured with IR Thermometer on a hot plate (give it +- 5 %)
Melting Point
83 - 88 °C
First fumes starting
190 °C
No fumes arising anymore
240 °C IR-Spectrum (measured in attenuated total reflection method (ATR) UV-Vis spectrum (concentration < 0,01 wt-% in Acetonitril) Bioassay:
no effect lol long story: 20 mg 5-AcO-DMT were vaporized over the course of 10 min. The vapor taste is very smooth and not yukky like 5-OH-DMT. Also it does not turn black upon heating and vaporizes without any residue - just like pure DMT. In contrast this is not the case for 5-OH-DMT. It quickly turns black above 170 °C and only 50 % will vaporize, while the other 50 % will form a black burned layer on the surface, no matter how soft and gently you are heating it. Seems like the 5-OH is the source of evil regarding the properties of 5-OH-DMT aka. Bufotenine which makes it hard to crystalize and also generates an unconvenient vaporization profile. Therefore the 5-AcO seems to make the molecule much easier to handle in any way. Nevertheless no effect was felt.  Vaporization overcomes first-pass-effect by the liver, maybe this is a reason. 5-AcO-DMT should be a prodrug of 5-OH-DMT. De-Acetylation should be fast in the body, but no idea where it takes place - maybe that step is skipped when inhaling vapors. Nevertheless that would mean the prodrug is totally inactive. Compared to Psilocin, its prodrug 4-AcO-DMT is reported to be instantly active when inhaled, suggesting it has an effect itself. Therefore it would be strange if thats not the case for 5-AcO-DMT. Only solution would be: What you see in the first picture is not 5-AcO-DMT, which is nearly impossible, but cant be proven by MS or NMR right now. Still, it has a distinct Tryptamine / Alkaloid taste. Maybe I should spend some time comparing the IR to this one. 3257 is hard to see, but might be there (2186 not in literature spectra) 1752 is there (1698 not in literature spectra - Acetic Acid?) 1373 is there 1216 is there 1168 is there 948 is there ... seems like true 5-AcO-DMT. 2186 + 1698 might be any residual stuff, but their signals are quite strong and in contrast the purity should be super high.
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Harmala Alkaloids Solubility:Many people have debated about Harmala Solubility and what to use if making Changa. Myself I had the feeling like they are soluble in exactly nothing. Here are the most common solvents, from good to bad. boiling DCM (40 °C) 24,9 g in 1 L 2,5 g in 100 ml hot DMSO (100 °C) 23,3 g in 1 L 2,3 g in 100 ml boiling Methanol (64 °C) 12,7 g in 1 L 1,3 g in 100 ml boiling IPA (82 °C) 8,2 g in 1 L 818 mg in 100 ml boiling Ethanol (78 °C) 8,0 g in 1 L 802 mg in 100 ml boiling Ethyl Acetate (77 °C) 7,9 g in 1 L 787 mg in 100 ml boiling Aceton (56 °C) 3,6 g in 1 L 360 mg in 100 ml Seems like from any good solvent DCM would still be the most healthy one. DMSO is totally healthy, but will never evaporate, so this does not help for Changa people. But if you can be sure to 1000 % evaporate stuff, then Methanol would also be an option. If being more on a safe side, Ethanol is then the best. It is also possible to just prepare everything in an even 1-layer and just sprinkle the Harmalas on top. Phase Transitions:Measured with IR Thermometer on a hot plate (give it +- 5 %) Melting Point / sublimating directly First fumes starting 180 °C Sublimation while forming small black clumps 205 °C No fumes arising anymore, leaving behind black residue 240 °C I think 225 °C is optimal for vaporization.Interesting, I thought it would be ways above DMT. That means that vaporizers like the Crafty / Mighty can indeed also partioally vaporize Harmalas, it was always stated they could only be used for infused herb. Maybe in future I may test Harmin and Harmalin on their own, for now only the mixture. But they should not differ too much anyways. IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (0,05 mg/ml in Acetonitril) In contrast to DMT we can see a slight absorption in the visible range (> 350 nm). Thats why Harmalas are not colorless. They have the same molecular size, but the chromophor has a slightly bigger pi-system. Thats because it's partially reduced and carries more pi-electrons and therefore the energy between lowest occupied orbital and highest non-occupied is smaller. This will shift the absorption towards lower energies aka. higher wavelengths. I bet that if you would measure it for ONLY Harmin and ONLY Harmalin you would see that the 350 nm+ absorption is caused by Harmin - as it has the biggest pi-system and should therefore be mostly causing the coloration. Thermogravimetric Analysis (TGA) (Heating rate = 10 K / min) Here is the thermogravimetric analysis of Harmala Alkaloids. Heating rate is 10 K / min. What you can see here is the mass loss of a sample while heating. Thats very nicely demonstrating any evaporation effects. So what you see here: First Vaporization starts at 192 °C and ends at 270 °C. Sadly we can see the same thing as with Bufotenine - very ineffective vaporization and most of the material will just undergo pyrolysis and form a black tar residue. At 250 °C only 25 % of the Harmalas evaporated even though the temperature program is very gentle and only 8 mg of sample have been used. So this once again shows smoking harmalas may not be the most rewarding ROA. Differential Scanning Calorimetry (DSC) (Heating rate = 10 K / min) Here is the differential scanning calorimetry analysis of Harmala Alkaloids. Heating rate is 10 K / min. What you can see here is the uptake or release of Enthalpy of the sample while heating. Thats very nicely demonstrating any phase transition effects like crystalization / melting. A rising graph indicates an endothermic process, which consumes energy and a decreasing graph vice-versa. So what you see here: Basically nothing hehe. Just confirming the Ghetto-Hot-Plate-Experiments there is no melting point and Harmalas sublimate directly. Ratio of Harmalin/Harmin This can be easily determined by 1H-NMR. The additional sp3-Protons of Harmalin give Signals at 3,8 ppm (t) / 3,97 ppm (t) both not being homotop, 2,73 ppm (t). 1H-NMR can be seen in this picture. Ratio is: 1,1:1 regarding Harmalin:Harmin at most, if not even more close to 1:1. No Vasicin can be seen. Additional tests were done using NMR during the separation of Harmaline and Harmine. See results here. Comments: More comments on this and the 1H-NMR can be read in this post at 8.). Bioassay:400 mg Harmala Alkaloids were eaten. Just a quick test to verify whether they indeed also contribute to Nausea. Intense vomiting. Nausea for hours. Really strong effect, like a mixture of LSD / stoned. Walking not easy. All in all bad experience. Close-Up: (Boring amorph powder - but you can re-x from DMSO to get crystals)
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Tetrahydroharmin 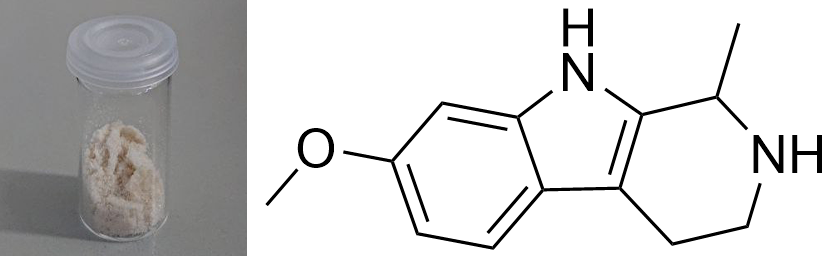 Solubility:Boiling Ethanol (78 °C) = 9,01 g in 100 ml = 90,0 g in 1 L Ethanol at RT (20 °C) = 3,76 g in 100 ml = 37,6 g in 1 L Ethanol in Freezer (-20 °C) = 2,39 g in 100 ml = 23,9 g in 1 L Insoluble in Naphtha Besides medium solubility in Room Temperature Acetone, IPA, Methanol, DCM. To separate it from contaminations of Harmin remaining from the synthesis, you can use Ethanol re-x - here are some more informations on this. Phase Transitions:Melting Point derived from DSC 188 °C onset 199 °C Peak maximum Literature: 187 - 190 °C Vaporization Temperature from TGA Starting from 190 °C+ IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (Acetontril, didnt write down concentration) Unlike Harmalas we can now see an Absorption dropoff just after 320 nm. In contrast Harmalas (and probably especially Harmine) absorb pretty weakly until 400 nm. Therefore when being pretty pure, THH is just white, as no absorption in the visible range is possible. Thermogravimetric Analysis (TGA) (Heating rate = 10 K / min) As seen above THH vaporizes much better than those Harmin / Harmalin. Unlike them you will vaporize it cleanly to a high degree, nearly as much as with the *real Tryptamines* like DMT / 5-MeO-DMT. Therefore it looks like you could also just pretty nicely smoke this one instead of just eating it. Probably efficiency will be just low, as you also need > 200 mg when eating it. Differential Scanning Calorimetry (DSC) (Heating rate = 10 K / min) Melting point onset for this THH sample is 188 °C, just the same when it starts to vaporize. Seems like you not really encounter any liquid THH as it will directly vaporize away. Still I would not call it a real sublimation as you still get a melting peak. Literature melting point is 187 - 190 °C, so quite in accordance. Bioassay:420 mg THH + 80 mg Harmin was eaten. At t + 1 h felt quite some sedation and a dreamy state. Lasted for at least 3 hours. But nothing like CEV or similar. Would compare to effect of 3 beer. Enjoyable for couch time, but nothing psychedelic. Read more here. THH can be vaped without any combustion - as opposed to Harmalas. 50 mg of THH was vaporized over the course of 10 min. But no effect was felt. Reason seems a very inefficient uptake of THH inside the lungs. Even after 15 sec of holding the breath most of the vapor just comes out when exhaling. Read more here. Comments: NMR-Spectrum can be seen here as a reference. Purity ~ 98 % with Traces of Harmin. Close-up: (it will always be amorph when precipitation at synthesis end - but can be crystalline with Ethanol re-x) 
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Salvinorin A (Salvia divinorum) 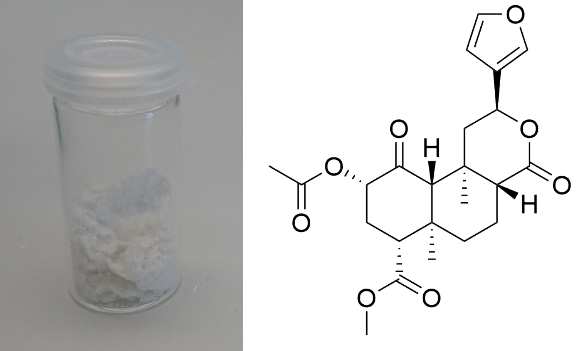 Solubility:Boiling Methanol (64 °C) > 80 mg in 10 ml > 800 mg in 100 ml RT Methanol (20 °C) 18 mg in 10 ml 180 mg in 100 ml Freezer Methanol (-20 °C) 3 mg in 10 ml 30 mg in 100 ml boiling IPA (82 °C) (slightly soluble, just try low amounts like multiple portions of 5 ml per 100 mg) IPA (RT) 0,74 mg in 1 ml due to Erowid1 mg in 1 ml due to another Erowid page, but cant find Aceton (RT) ~ 23 g in 1 L ~ 23 mg in 1 ml due to Loveall hereWater (RT) 12 mg in 1 L so basically 0 ... according to PubChemYou can create pure Salvinorin A fairly easy using this TEK or this TEK. Phase Transitions:Deleted hot-plate experiments with IR Thermometer - just directly check TGA / DSC. IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (2 mg/ml in Acetonitril) 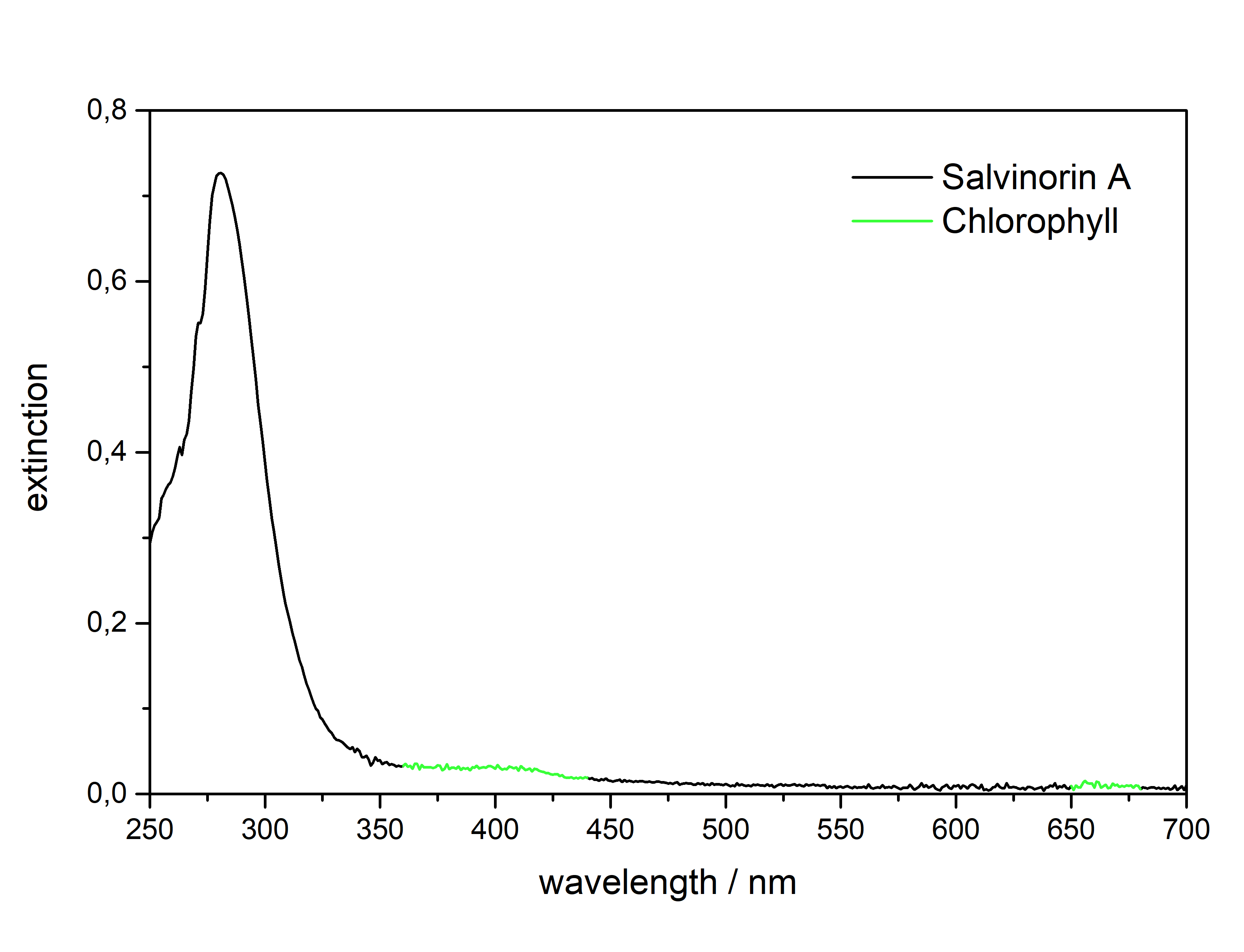 Even though this Sample was basically pure, even the slightest traces of Chlorophyll can be still seen at the both absorption ranges of ~ 400 nm and ~ 660 nm. Quite a proof how Chlorophyll is natures top player at catching light - even nanomolar concentrations will have a detectable absorption. Also it's an interesting demonstration why our world is green: If Chlorophyll absorbs at these 2 wavelengths, guess what the resulting light is: White - Blue - Red = Green. But apart from this that Spectrum is super boring, only an Absorption at 280 nm because of the Furan Ring. And this is also a weak one, as the concentration of this sample is 4-5x higher than of what I used for Tryptamines. Check how it looks with more Chlorophyll at the end of this post. (Heating rate = 10 K / min) 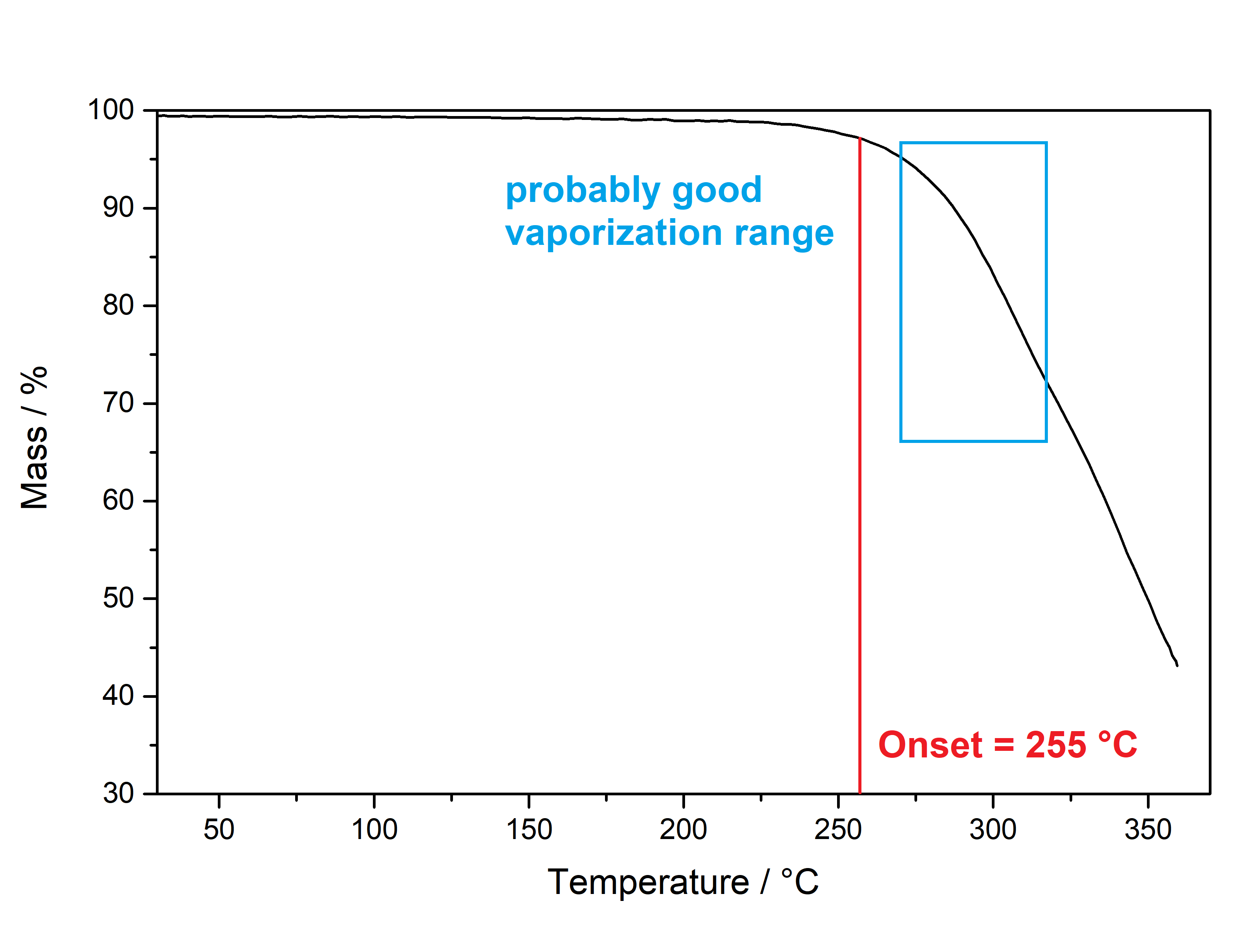 Here is the thermogravimetric analysis of Salvinorin A. Heating rate is 10 K / min. What you can see here is the mass loss of a sample while heating. Thats very nicely demonstrating any evaporation effects. So what you see here: Vaporization of Salvinorin A starts at ~ 255 °C. I am a little sad that I did not wrote down Test-to-400°C and instead used 350 °C. That means now we dont know if Salvinorin A evaporation could be complete like DMT or if it will decompose by pyrolysis at some point and you can never evaporate everything, just like Bufotenin or Harmalas. Just from the graph I would still assume mass drops probably still more than 38 % what we sere here as last point. Anyways it nicely shows that if you set a Vaporizer to 270 - 320 °C you can easily vaporize Salvinorin A. More than 100 °C above DMT optimal vaporization temp, but still quite low considering the size of that Molecule (M = 432 g/mol which is 230 % of DMT size). I read somewhere theory is that Salvinorin A itself never evaporates, but is transported in droplets to your lungs, which are formed when heating up the plant material. Obviously it still can just do the regular vaporization like our other favorite molecules. Also as you only need less than 1 mg for a Salvia experience, it may be that also lower temperatures can still evaporate enough traces. Loveall found even promising results at 180 °C! Update: TGA was only measured until 350 °C. See results up to 450 °C down. As seen after 350 °C there is a change in evaporation. Probably degradation starts and whatever product is formed does not vaporize so easy anymore. TGA RT -> 450 °C - click here. (Heating rate = 10 K / min) Here is the differential scanning calorimetry analysis of Salvinorin A. Heating rate is 10 K / min. What you can see here is the uptake or release of Enthalpy of the sample while heating. Thats very nicely demonstrating any phase transition effects like crystalization / melting. A rising graph indicates an endothermic process, which consumes energy and a decreasing graph vice-versa. So what you see here: Salvinorin A indeed has a Melting Point with a Max at T = 231 °C. Previously I could not observe it by eye, as my sample was contaminated with Chlorophyll which obviously will not melt. No idea what happens at 350 °C? That sudden increase in Energy Uptake indicates a strongly endothermic process, but based on the TGA there is no new effect observable. Not sure what that is ... But again I dont have much idea about TGA/DSC in general. Update: DSC was also only measured until 350 °C. See results up to 450 °C down. As seen at 350 °C there is a small endotherm process happening. Probably the degradation that also slows down the evaporation as seen in TGA. DSC RT -> 450 °C - click here. Salvinorin is a weird drug: It has no nitrogen, neither being a Monoamine nor even an Alkaloid. It is classified as Diterpenoid, a group of plant-derived components that originate from Isopren Polymerization. Also it does not interact with any of the 5-HT receptor family, instead κ-Opioid receptors. Therefore it is quite of an oddity in the world of psychedelics and anyways in Literature is is instead described as a Dissociative. Being a Diterpenoid it is also absorbing super weak, therefore naturally is invisible in a TLC and the absorption spectra looks super boring ... Still it is noteable that this big guy can be vaporized, according to the molecular mass I would assume it would rather decompose, but not having too many reactive species like double bonds or amines, this probably saves the molecule from disintegrating too early on heating. Anyways, that route is the only viable option as it is quite accepted that any other ROA (snorting / eating / quidding) requires other plant actives to make if bioavailable these ways, which are lost upon isolating the pure compound. Still I'm quite interested how a pure Salvinorin A + Cyclodextrin Nose Spray would work. Close-Up: (sharp colourless needles - ouch - see more crystals here) 
|
|
|

Boundary condition

Posts: 8617 Joined: 30-Aug-2008 Last visit: 30-May-2026 Location: square root of minus one
|
Great work as ever, BW!
I'm thinking of tabulating the data as you complete it. (As an aside, most of the chemical terminology can be anglicized by adding an -e on the end of the German term:
acetonitrile, ionophore, etc. but phenol and the -OH "-ols" stay the same. "Indol" becomes "indole", however, on account of its lack of an -OH group. Etheric -ols like "cineol", "anethol" and "safrol" take the -e, so to speak , and "-in" becomes "-ine" if it's nitrogenous: e.g. "anilin" → aniline, but salvinorin, for example, stays the same. And for some reason, psilocin, baeocystin and psilocybin are random exceptions to this rule - with bufotenin(e) seemingly randomly switching between both camps 
Anyhow, I think your bandwidth is best taken up doing what you do best but the footnote is here for those who may have been wondering about some of the chemistry terms.)
Updated data for mescaline salts presented here. All credits: Brennendes Wasser (plus anonymous co-workers?) mono-Mescaline Citrate 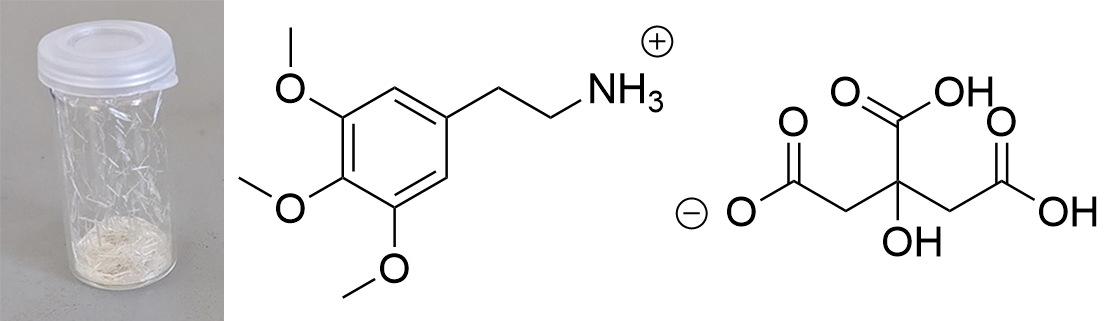 Solubility:boiling IPA (78°C)
3,5 g in 100 ml
350 mg in 10 ml
IPA in the freezer (-20°C)
530 mg in 100 ml
53 mg in 10 ml Mescaline can be extracted and solidified as a Freebase compound, but it will quickly react with CO2 and H2O from atmosphere to form Mescaline Carbonate. Hence all TEKs come with a way to produce the respective mescaline salt in the end, as storing it as a Freebase (like DMT) will not work. Regarding solubility a salt which is produced from a strong acid like HCl or H2SO4 will only dissolve in water or water:alcohol mixtures. CIELO as the go-to TEK for Mescaline will produce Mescaline Citrate, which is a salt from a weaker organic base and therefore it has a reasonable solubility even in pure alcohols (like EtOH / IPA). Therefore those can be used conveniently to recrystallize it, but obviously if you got some pretty crystals directly from the CIELO Tek this will probably not be mandatory  Besides very soluble in Room Temperature Water and DMSO. IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (0,6 mg/ml in Methanol, not Acetonitril) 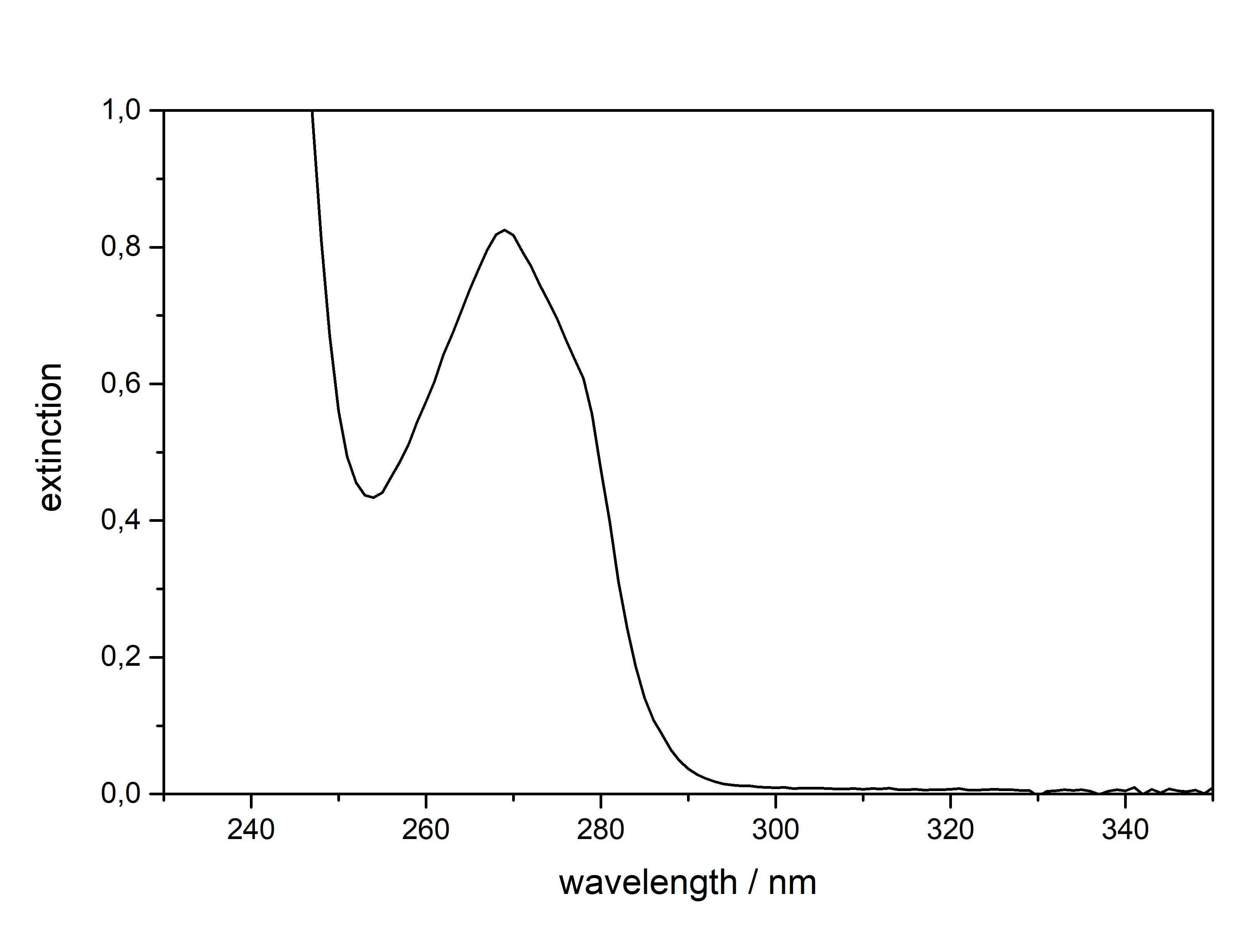 Note that Mescaline Citrate does not dissolve in Acetonitril in the concentration required, so Methanol had to be used. Now you may say this spectrum is kind of boring, not showing any absorption in the visible range (400 - 700 nm), thus this compound will simply be some white crystals, with at maximum some greyish tints from remaining Chlorophyll. But actually we could take the time to discuss a bit about photochemistry here, when comparing to (the structurally very different) other Tryptamines listed here. Both show an absorption maximum at 270 nm (Mescaline) and 280 nm (DMT). This is quite in the same range, Mescaline just being a little more narrow and *shifted left*, hence it needs a little more energy to show absorption.
The actual absorption spectrum or more precisely the position on the X-Axis of an absorption peak and also the width of the peak itself is determined by the molecular structure of a compound. Easily spoken, the more pi-electrons (AKA doublebonds) a molecule has, the more the peak is shifted to the right side AKA more likely that it will result in the molecule being colourful to us. This effect is much stronger if those double bonds are in close proximity, thus constructing aromatic ring systems. Now this effect is even more increased if we add more groups onto those rings, which will push and pull electrons across those rings.
Now after all that theory, we can see why all of these compounds have exactly those absorption spectra:
Mescaline has the smallest aromatic system of only 6 conjugated pi electrons. Its absorption spectrum is the furthest on the left side with the smallest width. Moving to DMT it has an additional conjugated 5 membered ring with 3 more conjugated pi electrons. Therefore the absorption maximum is shifted to the right and has a bigger width. Now attaching a Methoxy group (5-MeO-...) or Hydroxy group (Bufo) will further increase the peak width. When finally moving to Harmalas the pi system has the biggest size, therefore the absorption is so far extended that we start seeing this compound as being colourfull. This is also the reason why Mescaline in general has the weakest absorption: We have to use roughly the 2x - 3x concentration for the same absorption strength - Tryptamines usually only require 0,2-0,5 mg / ml. Thermogravimetric Analysis (TGA) (Heating rate = 10 K / min) 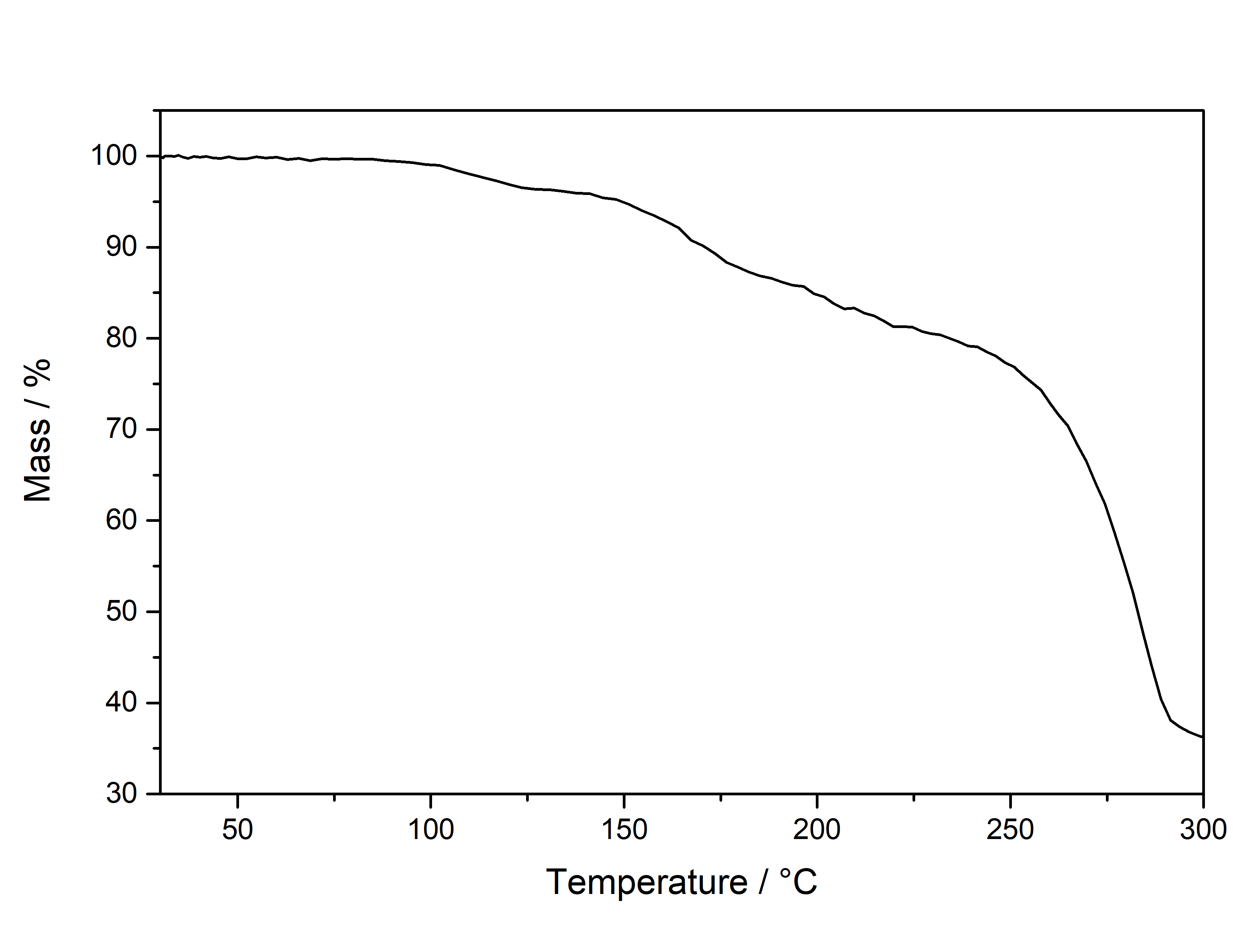 As stated before TGA shows the evaporation of a compound across an increasing temperature ramp. Normally you would say why measure the evaporation of something not intended for smoking, but well here we have it. Wikipedia tells that citric acid will undergo thermal degradation after 175°C. This is also what we probably can read from this data. First there is no clean evaporation, which would result in a slowly increasing slope and transitioning fast towards 0 % Mass (on Y-Axis). Instead we see some mass decrease even at lower temperatures before 150°C and then moving down to 40 % mass in plateaus. This indicates no full evaporation, which always is a good hint that a lot of product just ends up in pyrolysis and charred to a black mass, also the multiple plateaus may indicate a sequence of decomposition reactions. So in total you might not be able to vaporize this compound efficiently, though it would still be perfectly usable for nebulizing devices.
With some additional hotplate experiments I just observed formation of a black tar above 200°C with only minimal vapor release. Differential Scanning Calorimetry (DSC) (Heating rate = 10 K / min) As stated before DSC shows the occurence of energy releasing or uptaking processes like melting or decomposition across an increasing temperature ramp. Here we can see a melting point of Mescaline Citrate with an onset at 102°C and a max at 120°C. The peak is astonishingly broad and not very sharp as other Tryptamines. When observed by eye I could see myself that it first starts liquifying to a sticky blob at around 120°C and not forming a real molten liquid. This may correspond to the unusual melting peak seen in DSC. Still if there is no real reason vaporizing this salt it might not matter at all that it cant be formed into a true liquid easily. Comments: Mescaline Citrate can be conveniently collected as a product from the CIELO TEK and seems to be very pure even without purification. You may try an re-x in alcohols, but it is probably not worth the work if everything anyways crystallized well. Chlorophyll diluted just above the detection limit for the human eye will look greyish. In case your CIELO product has a faint colouration dont worry, because even the slightly coloured crystals from the picture above have 99,9+ % purity seen in HPLC. Close-Up: (The CIELO TEK will produce beautiful and delicate needles) Mescaline Benzoate  Solubility:
Toluene (20°C)
1,4 g in 100 ml
140 mg in 10 ml
Hot Limonene (150°C)
2,5 g in 100 ml
250 mg in 10 ml
Limonene (20°C)
500 mg in 100 ml
50 mg in 10 ml
Mescaline Benzoate has a mediocre solubility in non-polar solvents (which at least contain some double bonds) so those can be used to combine both Mescaline Freebase and Benzoic Acid and then precipitate the resulting salt. The solubility seems to be similar to the one of DMT Benzoate. Therefore if handling a big volume of Toluene, then chances are that you have too much liquid to start precipitation. Therefore it seems not feasible to extract with Toluene from the cactus, because you will end up with a too low concentration for crystal growth.
Furthermore this data is only valid if handling exactly the pure Mescaline Benzoate. If extracting from a cactus then the liquids will contain some more compounds, which might actually stabilize the Mescaline Benzoate in the mixture, therefore making it more soluble to a degree where it will even not precipitate if you concentrate evaporate the liquid even more. Therefore you will be only safe to use the numbers as shown above if you know that the Toluene and Limonene will not contain other substances.
Pure Benzoic Acid solubility at 20°C is 70 mg / 1 ml in Toluene and 47 mg / 1 ml in Limonene. Use these numbers to make sure that you dont go below that concentration, so any precipitation can only be your Mescaline Benzoate. Besides very soluble in Room Temperature Water and DMSO. Soluble in EtOH, IPA. Lower solubility in Ethyl Acetate. IR-Spectrum (measured in attenuated total reflection method (ATR)) UV-Vis spectrum (0,15 mg/ml in Acetonitril) Similar to the spectrum measured for Mescaline Citrate the shape of the absorption peak is not very interesting ... but we can see something else: In contrast to Mescaline Citrate we now have an acid anion which also will absorb light at 270 nm. Benzoic Acid has the same absorbing unit (simply being a phenyl ring) and thus both molecules of the salt will absorb at the same wavelength, increasing the absorption quite a lot. Therefore much less material was used to measure a similar absorption strength. I would have expected that the peak of Benzoic Acid would be a little more shifted left (because it lacks the 3x Methoxy groups). Still that seems not the case and we get exactly the same shape of the peak. Thermogravimetric Analysis (TGA) (Heating rate = 10 K / min) 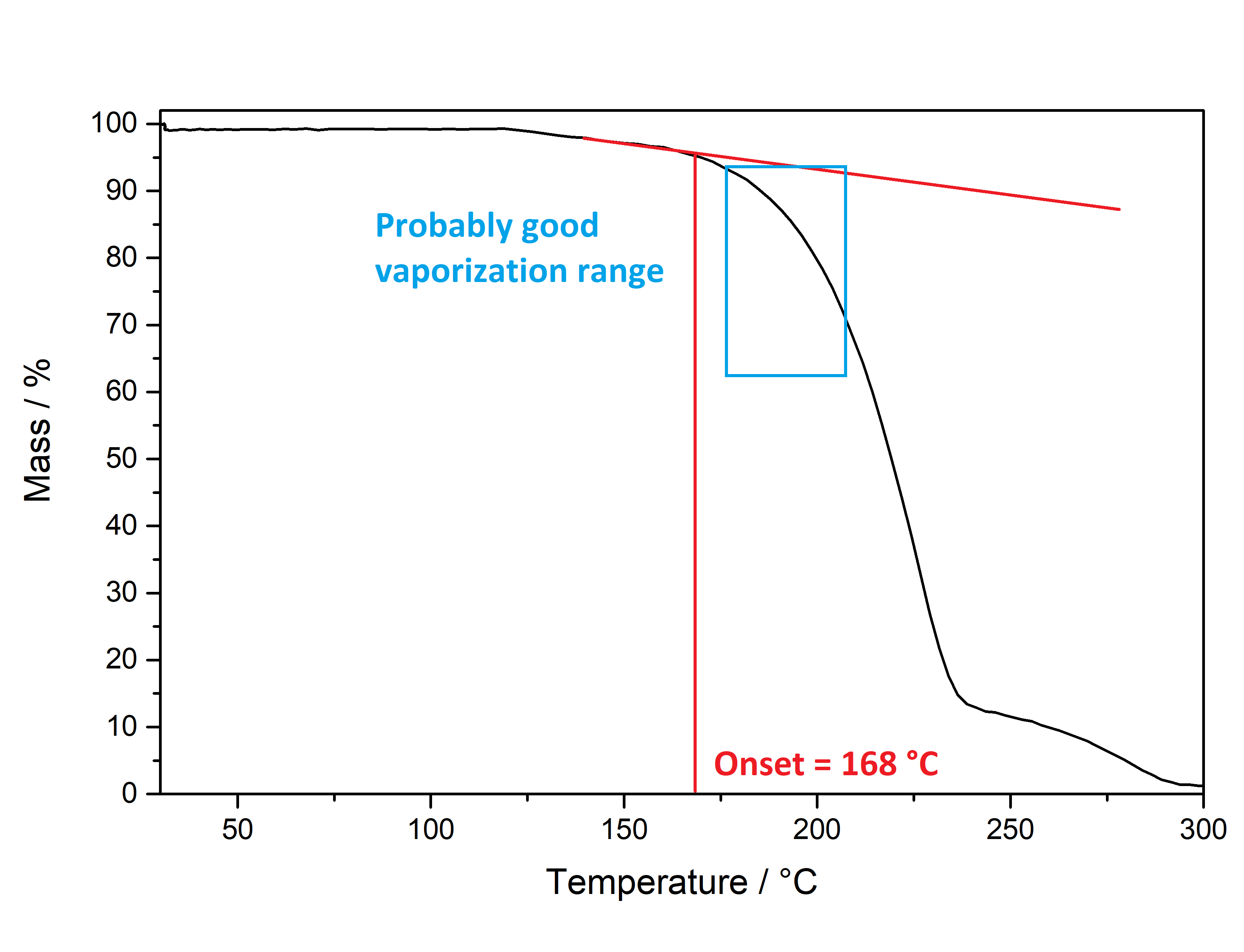 As stated before TGA shows the evaporation of a compound across an increasing temperature ramp. Prior we measured the TGA for Mescaline Citrate and it looked like it would mostly undergo pyrolysis instead of a clean evaporation. But as seen already for DMT and Bufotenin the salt created with Benzoic Acid seems to also evaporate very cleanly and efficient. This might be caused as for true vaporization only the nucleation of small droplets in air is mandatory, which will drag up all the liquid in form of vapor over time - read more about this here. Therefore salts created from Benzoic Acid vaporize basically like their Freebase analogues. This was tested here and indeed the vaporization courve is very pretty! It shows a very harmonic drop starting at around 168°C and the vaporization stops at first just above 10 % before 250°C, so it can be evaporated very clean. Afterwards there is a small further drop to nearly 0 %, but no idea what this unusual final part is, anyways the vaporization curve looks perfect for vaporizing this salt. Indeed I tried it and it works very smooth and clean, read more about this here. Still obviously it is a debate if smoking Mescaline is worth it, as it is in general a "weak drug" due to the very fast metabolism inherited by its chemical structure. I tested it only with 25 mg Freebase Mescaline Equivalent and the effect did not include any OEVs / CEVs. (Heating rate = 10 K / min) 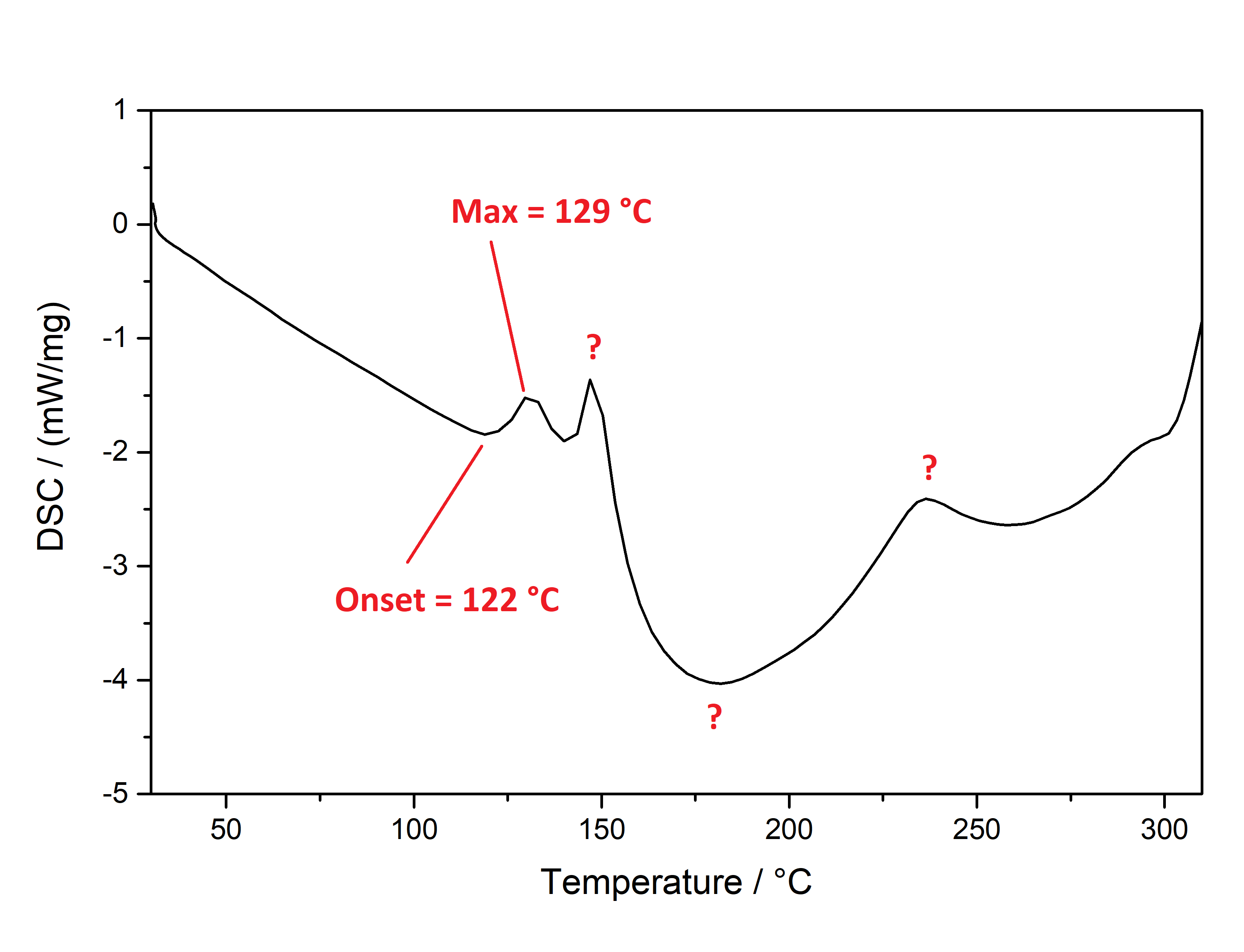 As stated before DSC shows the occurence of energy releasing or uptaking processes like melting or decomposition across an increasing temperature ramp. Luckily I will go pretty sure with the excuse that I dont have much knowledge in DSC and therefore my wisdom is simply just enough to always write those short paragraphs at each substance. Based on that looking on this spectrum I have no idea why it has this weird shape. Pretty safely it shows a melting point with an onset at 122°C and a max at 129 °C. This also corresponds pretty nicely with salts from other organic acids, which also were measured between 120 - 170°C. Mescaline freebase as a pure compound is melting very low at just above 30°C IIRC, so it's probably reasonable that the -Benzoate salt melts a little lower than the DMT-Benzoate. But besides that you first see a new maximum at around 150°C. That is weird because there should not be 2 melting points. Later you can again see a new maximum at around 230°C. Also this whole curve has a weird slope upwards. Therefore I happily tell I am a noobie in this thermal analysis science, so maybe a process is happening which other people might explain at some point xD. Comments: Mescaline Benzoate is an alternative salt with interesting properties, as it can vaporize pretty cleanly and also will form very delicate and beautiful crystals, with a shape seemingly strongly directed by the benzoic acid itself. It is very non-hygroscopic and forms voluminous crystals, so it is also very convenient to handle. Still it seems that there are no easy ways to directly produce it from a usual extraction TEK as solubility in most solvents is still too high. On top of that vaporizing this compound might be interesting, but it has to be proven that it can hit strong enough to make it worth more trials. Mescaline Benzoate can be created conveniently from Mescaline Citrate. Just use this TEK and find info of how you can smoke it, if desired. Bioassay: 40 mg Mescaline Benzoate was vaporized at ~ 190°C. Quite a strong effect on the body was felt. Warm and pleasant feeling. Still this only translates to 25 mg pure Mescaline which would be regarded less than 10 % of a common dose if taken orally. More experiments might be interesting, also vaping this while having a MAOI effect. Close-Up: (Super long needles like pure Benzoic Acid - micro droplets of Limonene still visible at right picture) [Once again, countless thanks to Brennendes Wasser for his amazing efforts!] “There is a way of manipulating matter and energy so as to produce what modern scientists call 'a field of force'. The field acts on the observer and puts him in a privileged position vis-à-vis the universe. From this position he has access to the realities which are ordinarily hidden from us by time and space, matter and energy. This is what we call the Great Work." ― Jacques Bergier, quoting Fulcanelli
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Summary of all evaporation experiments visualized in just one diagram: Interestingly there seems to be a pattern of quite some Alkaloids regarding their evaporation. Read about this here. Summary: Substances that are easily vaporized at 170-190 °C:DMT DMT-Benzoate 5-MeO-DMT (probably also 5-MeO-DMT Benzoate) Substances that are easily vaporized at 190 - 230 °C:THH Substances that are partially vaporized at > 230 °C and mostly decompose:Harmalas Bufotenin Substances that are easily vaporized at > 280 °C:Salvinorin A, still it degrades after 350 °C probably as seen here. Harmalas and Bufotenin are quite bad for vaping, if you care about efficiency: Most of them are just charred to death and leave behind a black residue. You will destroy actually more molecules than actually vape them  Salvinorin needs the highest temperature, but that comes to no wonder - it's just gigantic. Sadly the graph was not measured to 400 °C so we don't know it is also charres down. My opinition: I'm highly convinced it will vaporize without too much combustion, because it does not have any (strong) Nucleophiles or double bonds which are prone to Addition/Polymerization that would react at high temperatures to give non-evaporating Dimers/Oligomers. Summary of 1H NMR spectra of various compounds: Take care that the atom counting is kind of random and chosen by the software. So same atom may have another digit across similar compounds.
|
|
|

DMT-Nexus member
Posts: 14191 Joined: 19-Feb-2008 Last visit: 20-Jun-2026 Location: Jungle
|
Thanks for the experiments and sharing of data! When this is completed, we can update the wiki to add such data. Great work friend 
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Now DMT- N-Oxide added. In short: When handled pure and in the correct way its not a red oil, but a light-orange waxy solid. Furthermore when heating that stuff it decomposes even on Quarz Glass already just above 100 °C, making it nearly impossible to ingest by smoking. But even when eaten there is no effect felt. At least it tastes like Cherries! So I think that despite people calling the N-Oxide active, they probably never smoked the real N-Oxide, instead just real DMT with at maximum traces of it, as much as I think the *Jungle* experience from Jungle Spice just arises from the still majority of DMT. Therefore I think this stuff is near-safe inactive. PS: Now had to correct something, N-Oxide is not only soluble in HCl, but also just plain water. Also I forgot the maybe the most important thing of the Bioassay: It tastes like Cherries! So it has that slightly spicy ginger-like burning taste from regular Tryptamines, but it adds this cherry flavor, making it tasting quite like these Chewing Gums .
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Some new data added:
Differential Scanning Calorimetry (DSC)
and
Thermogravimetric Analysis (TGA) But only for DMT, as this may be the most important one This does not show any ground-breaking new stuff, but havent seen it elsewhere. It's basically a way of investigating when the sample melts and vaporizes. Looks quite like what we know, but now thats some different way of taking a closer look.
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
More data added: Bufotenin and Harmalas now complete. Summary: Bufotenin - slightly adjusted solubility in Xylene - UV/Vis added, it absorbs a little further than DMT because of 5-OH - TGA added, only 50 % will vaporize and rest does pyrolysis - DSC added, melting point ~132 °C - Closeup picture looks like vegetables (DMT also added, looking like Flowers as seen before) Harmalas - UV/Vis added, slightly absorbs in Vis-region in contrast to Tryptamines - TGA added, only 25 % will vaporize and rest does pyrolysis - DSC added, no melting point observed as before
|
|
|

❤️🔥

Posts: 3648 Joined: 11-Mar-2017 Last visit: 01-Jul-2026 Location: 🌎
|
Brennendes Wasser wrote:DMT-N-Oxide Synthesis:Dispense 1 g of DMT in 2,4 ml of 30 % H2O2 (does not dissolve as this is basically water). Add amounts of Ethanol while stirring until all DMT is dissolved. Should be ~ 7 ml in total. Stirr this mixture for 48 h at Room Temperature until full conversion. Evaporate all solvent, leaving behind DMT- N-Oxide. ( source) TLC after 48 h still showed a spot in height of starting material. Nevertheless the final product was extracted with boiling Hexane and this Hexane showed no clouds when Trifluoroacetic acid was dropped inside, indicating no residual Freebase so conversion was quantitative. Also the majority of solvent was evaporated by N2-stream. The last bit was evaporated on a hot plate ( Picture 1). This should be done with caution, as it was discovered the N-Oxide is very heat sensitive and decomposes already above 110 °C ( Picture 2, turning completely black just within less than 3 seconds). If treaten with care it will form a waxy orange solid ( Picture 3). Solubility:boiling n-Hexane (69 °C) insoluble Aceton (RT) insoluble DMF (RT) insoluble DMSO (RT) soluble Water (RT) soluble Wow quite bad solubility overall ... would have guessed it would be soluble in Aceton, but at least at RT there is no dissolving process to be watched. Still its soluble in even neutral water, given the properties of being a salt. Phase Transitions:Interestingly pure DMT- N-Oxide is an orange solid and not an oil contrary to popular believe. The oily consistence only arises due to always encounter it in regular ways in a mixture with DMT. Already small mixtures of substances make them not only prevent to crystalize, but also often forming an oil - this sample for example is an oil, yet 80 % DMT purity - even though they may both be an actual solid when being pure. Measured with IR Thermometer on a hot plate (give it +- 5 %) Melting Point 40-50 °C hard to determine due to waxy structure DMT- N-Oxide turning black 110 °C First fumes starting 130 °C No fumes arising anymore, black tar residue 150 °C I think there is no optimal temperature for vaporization. It just decomposes super fast, even on inert surfaces like glass. It does not even give strong fumes, it mostly turns into black tar. IR-Spectrum (measured in attenuated total reflection method (ATR)) DMT-N-Oxide separation:As the N-Oxide seems to be only soluble in DMSO, this can be achieved to do a purification from other possible tryptamine compounds. Yet it seems this can turn oily stuff into more solid products, but yet I would not call this an actuall recrystallization.
1. Dissolve the N-Oxide in a minimal amount of DMSO
2. Add 20x Volumes of Ethyl Acetate (possibly IPA could work, too)
3. The solution turns cloudy, place in the fridge for 3 h
4. Decant the clear liquid to retrieve your N-Oxide (Picture below). All non-oxidized Amines will stay in solution, no matter what Tryptamines they may be. Comments: So interestingly DMT- N-Oxide is a solid, but thats also what you can observe with DMT: Just having small traces of impurities it will already start to become oily. Still, the substance is quite waxy. Also interesting, but annoying seems the super fast thermal decomposition, starting from 110 °C and being done before 150 °C. This makes it super unlikely that anyone ever has smoked some real N-Oxide, also it seemingly being non-soluble in Acetone as it seems, preventing it to be transfered further into Changa. People still dissolving all their oily DMT in Acetone probably had no N-Oxide pollution, but just oily DMT due to other impurities. Actually I even think having any reasonable about of the N-Oxide in your extracted stuff is just super unlikely. Why should it have become oxidized? You would need a real oxidation agent or UV-rays and pure heat does not oxidize DMT - even when in direct contact with O2. Also UV-Rays will not make a full conversion. Therefore even if there were traces of N-Oxide in a plant-derived sample, they will always come with a big bunch of non-oxidited DMT. Only a real chemical conversion would lead to 100 % N-Oxide. So based on smoking oily residues labelled as N-Oxide and being called active, this may have just been a false positive thing. I more have the feeling that people in general call oily DMT being polluted by N-Oxide, but also even small amounts of pollutants will turn your goodies to an oil, this sample is a classic oily mess and still 80 % of DMT Purity. In regard to bioactivity of DMT- N-Oxide I think its nearly impossible to smoke this compound, as it will turn black within less than 3 seconds once heated above 100 °C. By that time it will not even have evaded any fumes to inhale. Further heating does just produce traces rising up from that super uggly and unhealthy looking black tar (see Picture 2). But as the N-Oxide is the natural product of MAO-enzymes, it should not be needed to block these when incorporating the N-Oxide. This means if it HAD any psychedelic effect one could simply eat any amount of N-Oxide and would suspect an effect not too far off from the same amount of DMT, eaten with 150+ mg Harmalas. Now people could argument, eating would be an inefficient ROA, as the high-polarity compound is not likely to tresspass membranes and will have low bioavailability. Actually it is even less polar than DMT, when eaten orally and DMT is indeed bioavailable when eaten, as people know. The proof can be seen here in this reversed-phase Chromatogram. The solvent protonates any bases, causing them to have uniform charging across the column. This happens to DMT and the N-Oxide, while now being DMT+H(+) more polar than DMT-N-OH(+). In a consequence, the N-Oxide elutes 1 min later in reversed-phase and the solvent can be transferred to our gastric conditions, where also both compounds will form their protonated salts. This way I am very certain, that if N-Oxide HAD an effect it will be visible upon oral ingestion. 80 mg of DMT- N-Oxide was eaten. No effect But: It tastes like DMT (Ginger-like spicy) + an additional Cherry Flavor. Super cool! Weird sweet bonus, exactly like Cherry Menthol gums. So while the N-Oxide being a metabolist of DMT and definetly has a metabolic pathway in the body, I highly doubt it would still interact with the same receptors that are also responsible for DMT-effects. That unoxidized Amine would normally be important for in-vivo recognition at receptors, rendering it possibly inactive upon oxidation, as the N-Oxide is a very different chemical species - unlikely to show the same binding behaviour. That said and given the facts that it cant really evaporated and shows no effect upon ingestion, I highly doubt that anybody ever was high on N-Oxide . I agree that we don't encounter DMT-N-Oxide, at least I have never come across it. The orange oils are mostly DMT. I think even pure DMT can take an oily form since it is a polymorph. Simply melt and cool to convert it from xtal to oil. Also, during the water basing step, if DMT is concentrated and in a high ionic strength solution, some FB DMT can polymerize in the water and come out as an oil during extraction. I think it is a good idea to keep ionic strength below ~10%, not have the DMT too concentrated, and pull quickly when basing or better yet have naphtha present while adding NaOH). I believe the max ion Tek is a good example of avoiding DMT polymerization. The DMT oil takes a longer to dissolve in hot naphtha. I fear a lot of good DMT has been thrown away during re-X. Next time anyone is chasing xtals and has a leftover oil from re-X, perform and A/B on it, xtals should appear. Another potential waste of good DMT is people not using warm naphtha because that gives oil instead of crystals. That could be just leaving perfectly active polymerized DMT behind. There are claims on the nexus of converting DMT-N-Oxide into DMT using metal dust and acid. I think that is not correct. They were simply breaking up the DMT oil polymorph with the acid, (the metal dust was not needed) and getting the DMT xtaline form. I could be wrong, but that is what all my experiments are telling me. By the way, thank you for this work Brennendes Wasser.
|
|
|
DMT-Nexus member
Posts: 147 Joined: 20-Jun-2021 Last visit: 14-Feb-2024 Location: Earth One
|
You are a beautiful alchemist Brennendes! Regarding acetateDMT, here`s a quote: Quote:Maybe it may be explainable by counting Acetate as a soft ion and not a hard ion like chloride or sulfate. Can you expound some of your wisdom regarding vaping DMT salts? Is there any prediction on which salt specifically would provide lower vaporization temp compared with fumaric DMT? Yes, salts vaporize poorly, perhaps there is a simple solution to that and your data may provide some guide into it (nicotine benzoic or levulinic salts recently surged into the vaping market as an example).
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Quote:Can you expound some of your wisdom regarding vaping DMT salts? Is there any prediction on which salt specifically would provide lower vaporization temp compared with fumaric DMT? Actually I dont even know why the DMT-Acetate does vaporize and not even feel bad to your lungs. But just following logics, I would use any acid with a Molecular Weight as small as possible. So Vitamin C / Benzoic Acid / Tartaric Acid will all be way heavier than Acetic Acid, so I would naturally simply start with the last one before investigating any other ... If you would do an aerosol based inhalation (like some people tried to investigate here years ago) it might not make a difference, but choosing an acid favored by pharmacy might be the best option regarding physical uptake, then I would go with Benzoe or Tartaric.
|
|
|
DMT-Nexus member
Posts: 823 Joined: 23-Sep-2017 Last visit: 05-Feb-2024
|
Now also THH added. Summary: Quite the same as Harmalas of course, but doesnt stop vaporizing after 250 °C. Maybe some kind of pathway of thermal combustion / polymerization is blocked here, enabling further evaporation. That may make the question interesting how vaping pure THH might be, if it might then even be a really strong uplifer, compared to the more gentle feeling upon oral ingestion.
|