Extraction of Bufotenine (5-OH-DMT) from Anadenanthera colubrina
It seems this TEK could be made shorter, but might require some validation. But you might still try the shorter route, which will directly give Bufotenine Benzoate, which is not combusting like Freebase Bufotenin and therefore much better to smoke. Check here.This TEK will produce pure Bufotenine from Anadenanthera
colubrina seeds, using a minimal amount of steps and non-toxic chemicals. This TEK was developed based on all the 37 pages of the other 2 sticky threads and combining the most useful methods, but it adds some tweaks and new ways and therefore will finally lead to a solid, crystaline product. In 3 steps all compounds with lower polarity will be removed, then all
fats will be divided from alkaloids, then all more polar alkaloids will be removed to give pure Bufotenine.
In the end there is a section of a GC/NMR-Analysis to prove purity and validate the effectivity of that TEK.
The first post is in detail with pictures and the second post is a short version. If any pictures are offline, you can download them at the end of the post with a .RAR file.
For general information about Bufotenin click
here.
For a chemical look into the harsh Bufotenin smoke and how to make it more pleasant click
here.
What do you need?- 100 g of Anadenanthera
colubrina seeds ("
Vilca", "
Cebil" )
1 Seed ~ 180 mg, 100 g ~ 555 seeds
- 1200 ml dry Acetone
(put 50 g of
anhydrous MgSO4 or Na2SO4 into Acetone and let it sit 1 night)
- 1000 ml Naphtha
(preferred: Hexane/Heptane isomers, "C6-C7-Alkanes", boiling Range 60 - 80 °C)
- 100 ml Xylene (if you want to evade Xylene you can use the Ethyl Acetate-Naphtha-Mix. See solubility table for how much solvent to use then)
- 400 ml of FASA, Fumaric Acid
can be replaced with Citric Acid from a Grocery or Benzoic Acid (for Benzoic you must evaporate Aceton down to < 50 ml before adding the BASA = benzoic acid saturated Aceton)
(prepared from your 1200 ml dry Acetone, according to
this source 0,92 g Fumaric Acids dissolves in 100 ml at 20 °C and 1,7 g at 50 °C)
- 1 package of soda/Na2CO3
- Cheap kitchen grinding machine like
this - a coffe grinder may also work
- do this in a country where this is legal
- Yield ~ 1-3 %
1. Preparation of the seedsPrepare your dry Acetone + FASA before starting this TEK to save time. Lay out 100 g of your seeds in a pan and turn the pan on the hottest setting. After the first seed pops open, turn the heat down to the lowest setting where you still notice more seeds popping. This is the sound of pressure building up and breaking apart the seeds, while volatile components leave the seeds. The smell is like tasty caramelized nuts, but dont forget to turn the heat to the lowest possible setting, or it will smell burned and all Alkaloids get destroyed. When you dont hear any more seeds popping, this step is completed.
This defat step will remove ~ 6 % of unwanted compounds aka. fats and/or water.Important: If fumes visibly arise then you already know that it is too high, as the release of those contaminations wont form visible fumes.
Important: place a cover on your pan, but dont close it completely or else the heat will quickly rise up inside of your pan and still destroy your seeds on the lowest heat (depending on your stove, again)
Picture of the seeds before (left) and after (right) cracking at low heat.
2. Freebasing the AlkaloidsNow place your popped seeds in the milling machine and create a fine dust. Then add 25 g of sodium carbonate (Na
2CO3, but not Na
HCO3 which is too weak) aka. Soda (1:4 to original seed weight) and mix both to a homogeneous powder. Now add water to the dry mass until you reach a dough-like consistency. While doing this a strong freebase-smell will develop.
Optimal ratio: 1,5 ml : 1 g original seed weight, which means 150 ml on 100 g Seeds.
Important: If you use Hydroxides like Lime (Ca(OH)2) or Sodium Hydroxide (NaOH) it may destroy the Bufotenine at high pH ranges from 12 onwards due to the irreversible oxidation of the 5-OH-Group to an Enone. Carbonate can only raise the pH to ~ 11 and should be used without exceptions.
Picture of the grinded seeds (left) and the mixture with Na2CO3 (right).
Let this mixture sit for 60 minutes and mix it from time to time. Afterwards the mixture needs to be dried. Either put it in the oven at 75 °C and turn on maximum ventilation or lay it out in a pan, turn heat on a LOW setting and use a fan from the side - this is much faster than the oven method. But heavily check temperature to not destroy any actives. Fumes should not arise, these indicate decomposition. The paste will shrink in size and form blocks. Crack them up to increase surface area and accelerate water removal. At the end use the milling machine again to form a fine powder again - greatly improves the extraction step!
Important: Note the weight of your
dry mixture so you know at the next step when you have removed all the water.
Picture of the seed paste (left) and the seed paste while drying in a pan (right).
3. Defatting the Alkaloids with NaphthaThis step is used to remove all the unwanted alkaloids aka
fats that are less polar than Bufotenine. Place your freebased seed/soda mix in a container and add 150 ml of Naphtha. Boil the mixture at 60-65 °C for 5 minutes in the Naphtha while preferably stirring - no need to be exact with temperature, just make sure that it is boiling. Wait for the seed powder to sediment down to the bottom, also shake the vial gently to make the paste form a solid packed layer. If the layer is very dense you can decant the Naphtha without spilling over any seed paste. Repeat 3x, better be safe, Naphtha is cheap and this step is essential.
This defat step will remove ~ 8 % of unwanted compounds aka. fats.In theory this step would contain all the DMT or 5-MeO-DMT from the seeds. By applying FASA to the Naphtha no clouding can be observed. It seems either it evaporated at step 1 or there is indeed nearly none of these in the seeds. You may check yourself.
Important: As you may not fully prevent spilling over seed material you may filter the Naphtha through a sneeze tissue while decanting.
Picture of the seed mixture while defatting with Naphtha (left). Clearly watch the dense package of the seeds, which will not spill over. Shake the vial smoothly to achieve this density. Picture of the setup to recover spilled over material (right).
Important: After this step the seed powder is basically the same as the traditional Yopo / Cebil / Vilca Snuff used by south american shamans for their visionary effects. I strongly recommend
NOT using them as a snuff if you used an electric milling machine, as this produces particles ranging down to extremely fine dust which WILL also get dragged into your lungs when insufflating. As they are not soluble in your lungs it may cause the same unhealthy effects like any other fine dust sources.
4. Extracting the Alkaloids with AcetoneNow the target Alkaloids are extracted from the freebased seed powder. Place them in the same container from step 3 again and pour 300 ml (to 100 g seeds) of your
dry Acetone on the powder. Boil the seeds at ~ 60 °C for 10 minutes in the Acetone while preferably stirring - no need to be exact with temperature, just make sure that it is boiling. Wait for the seed powder to sediment down to the bottom, decant the Acetone and repeat 2x times with fresh solvent.
Again, shake the vial gently before decanting, this will solidity the mass and make life much easier.
This extraction step dissolves 8 % of alkaloids and fats from the seeds (Bufotenine will be ~ 1-2 % of these).Important: Close the container while heating, but dont let pressure build up. You may use cling film and a rubber band to prevent air (and therefore water) to get inside of your dried acetone. Huge water take-up would reduce the yield later on.
Picture of the seed mixture while extracting with Acetone. You can see the solid material settled down for easy decanting.
Now to separate the Alkaloids from the other
fats FASA will be added to the solution.
(If you have Benzoic Acid by Hand you can also just use this.) By assuming that the Alkaloid content of the seeds is around 1-4 % (
reports of 13 % just sound unrealistic) you can expect 3 g of Fumaric Acid to be more than enough to fully precipitate every last Alkaloid. Add 300 ml from the 400 ml of FASA to your combined Acetone extracts.
This precipitation method will separate 3,75 g of Alkaloid Fumarates from the Acetone Extract, which
equals equals ~ 2,4 - 2,9 % of Alkaloids extracted, depending on being Mono- or Disalt Fumarates. (Bufotenine will be ~ 1-2 % of these)
GIF showing the addition of FASA to the Acetone Extracts.
Important: Even though the clouding is very intense it takes many hours for full phase separation and crystalization of the Alkaloid-Fumarates. So close the container and let the solution sit over night (at the stage of being fumarate salts the alkaloids have a very long shelf life just like any other fumarate alkaloids). Pay attention that even after full crystalization the solution will not turn clear, so dont wait for this moment. To be safe, decant it and keep the liquid to check if more precipitation is forming while proceeding the next steps with the Fumarate-crystals.
Important: Fumaric Acid + Acetone can be replaced by Citric Acid + Acetone, as Citric Acid can be bought from any grocery. Instead it will form a blob then, which looks like
this and will settle down immediately instead of having to wait. But it will contain a lot of Acetone, which will slowly drip out. Wait for 2 h while tilting the blob in a closed vessle, so Acetone can rinse out, while no water can stick to the Alkaloid-Citrates - they are quite hygroscopic in contrast to Fumarates.
Picture of the Alkaloid Fumarates after 10 h of precipitation (left) and in close-up (right). Looking nice  The big cucumber strucure is a stirring bar, in case you wondered.
The big cucumber strucure is a stirring bar, in case you wondered. Important: Dont rinse the Alkaloid Fumarates with Acetone. This will break up the crystals partially and you cant decant the Acetone without loosing too many material. Still, it is not even needed for purity.
5. Extracting Bufotenine from the Alkaloid mixtureDissolve your Alkaloid-Fumarates in 25 ml of hot water. Discard what does not dissolve. Then create a solution of soda/Na2CO3 in 25 ml hot water, use a ratio of 0,5:1 Na2CO3:Alkaloid-Fumarates. You should not use excessive Na2CO3, as Bufotenine may dissolve in water if you used too much Na2CO3. Decant to get rid of any non-dissolved soda. Slowly pour the Alkaloid-Fumarate solution into the soda solution. You should use a vial for the soda solution that you can use for heating in the next step.
You will recognize brown clouds of freebase Alkaloids form and settle to the bottom after a few seconds. Upon stirring they form a big clumpy ball.
GIF of the addition of Alkaloid Fumarates into saturated Na2CO3-solution. At the end you can see the brown freebase clump that is rolled in circles through the glass vial.
Now the Alkaloids of the seeds are reverted back to their Freebase form and can be extracted with an unpolar solvent. Decant the water, all the Alkaloids are present in the brown sticky blob. Dont worry if the water is still light tan, it still does not contain any Bufotenine. Pour 25 ml of Xylene into the vessel and boil it while stirring strongly (T = 140 °C). Take care to be in a well-ventilated area. If you dont want to use Xylene, use the next mixture down at the solubility table, which would be a 1:3 Ethyl Acetate:Naphtha heated to 70 °C and use portions like 100 ml per pull, as solubility is much lower).
At the moment of creating this TEK I still used that EA:Naphtha mix, so if not otherwise stated, pictures are generated from using those solvents. Still Bufotenin is much better as it is more selective and precipitates all your product at room temperature again. Now while stirring the blob will form a round ball. If the defat step was very thoroughly, then it should solidify to a hard ball after ~ 1 minute, if not it may stay sticky. This wont be a problem, if so just continue as normal. But if it solidifies you will need to stop stirring and crush it into fine dust. This is also a sign that Bufotenin migrates into the solvent. After 5 minutes of stirring, wait for the dust to sediment to the bottom, then decant into a new container. Repeat with fresh 25 ml Xylene until you have the feeling that the powder does not get less anymore - you may weigh the vessel after every Xylene pull for that. A brown or even black solid residue will be left at the end.
Solubility of Bufotenine / 5-OH-DMT in Xylene:
boiling Xylene (140 °C)
43 g/l = 4,3 g in 100 ml
Xylene in Freezer (-20 °C)
- 20 °C = 0,1 g/l = 10 mg in 100 ml
Important: Xylene is highly flamable, so handle with care. Also Bufotenin slowly degrades at 140 °C. It will still be worth to do the Xylene pull as material will be more pure, but a little bit of Bufotenin will also be destroyed meanwhile. Therefore dont extend this extraction step to 20 minutes of constant 140 °C or more.
Also if you dont like Xylene, it can be replaced with the Ethyl Acetate:Naphtha mix, but that also needs to be heaten up to 70 °C boiling temperature. Use exactly that ratio as it is just correct to not catch up too much unwanted actives, while still dissolving Bufotenin. Nevertheless it will not drop all Bufotenin when it freeze-precipitated, so instead you need to evaporate the solvent off at the end, which may give amorph instead of crystalline material. Still I also got crystals from this and never sticky oil if followed closely to the TEK. If using that mixture, also make sure to place a lid to the container to avoid too much evaporation, which would change the ratio of EA:Naphtha and hence the solubility.
Picture of the sticky Freebase Blob after decanting the water (left) and after crushing the solidified freebase blob to fine dust (right). Dont worry if it not solidifies, then the defat step may have not been 100 % efficient. Proceed as normal.
Now place all the collected solvent pulls in a fridge. If you used the EA:Naphtha mix then I suggest only combining the first few pulls as they will be the most saturated. Adding many less-saturated pulls to them will decrease the chance of recovering most of your material when doing a freeze-precipitation. After ~ 5 h you will see yellow to white either amorph or even crystaline solids on the bottom. Decant solvent and if using the EA:Naphtha mix you can evaporate the solvent too, to get another less-pure fraction.
Combined collected material = 1,13 g Freebase Bufotenine (1,13 % yield)
[later extraction: 2,6 g from 75 g Seeds = 3,4 % yield!].
Congratulations: You got yourself some solid pure freebase Bufotenine!
Picture of crystaline freebase Bufotenine, directly derived from the second pull of this TEK.
Picture of amorph freebase Bufotenine, directly derived from the first pull of this TEK.
Alternative Workup
You could also add FASA or Benzoic Acid and drop Bufotenine Fumarate as perfectly white crystals (next picture, left). These cant be vaporized, but you may use them for oral consumption. Regarding nasal administration it was reported that Fumarates are far less active, maybe due to the high polarity the bio availability is strongly reduced.
Also you may recrystalize the amorph Bufotenine. For this you should use Xylene and not Ethyl Acetate as mentioned in Literature. Using the latter one will turn the Bufotenin brown (next picture, right). So for recrystalization stick to the pictoral in Post #2. Also regarding solubility, also check post #2 or
here.
Picture of Bufotenine Fumarate generated from the remaining 1:3 EA:Naphtha solvent mix (left) and recrystalized Bufotenine (right) derived from the tan amorph Bufotenine. You can clearly see a darkening and these crystals turn dark, the non-recrystallized does not. Using Xylene for re-x causes no coloration.
Analytic data to verify purity of the crude Bufotenine
Bufotenine derived from this TEK was measured by Gaschromatography and 1H-/13C-NMR. By intention a non-crystaline, amorph sample was used, as it was believed to have a lower purity than the crystaline fraction. Therefore this should help to verify if even the lowest purity outcome is already of good quality and to check if the crude products needs a workup at all if the TEK is conducted correctly. Actually it seems the person did forget to measure the 13C-NMR, maybe I will deploy it in future, when I get NMR-credentials again. Still, its pure Bufotenine for sure, which you will already see in the following data, so 13C-NMR would be rather cosmetic.
At first here is the predicted 1H-NMR of Bufotenine, this is used to get an idea about the magnitude of impurities in the sample measured below - simulated with Mestrenova 12.
Picture of the simulated 1H-NMR-spectra of Bufotenine - from Mestrenova 12.
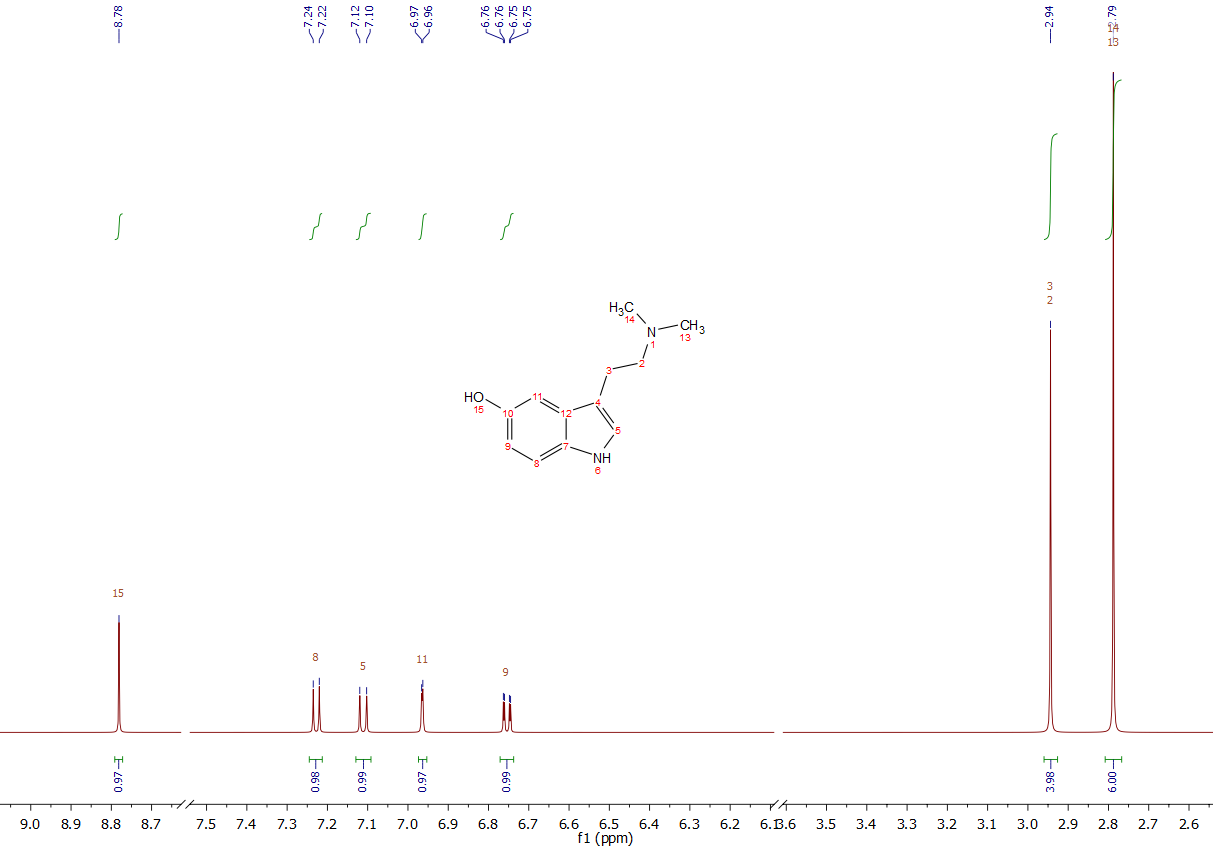
You can see, it looks practically like DMT, with a big 2x Methyl-Signal on the right and the 2x 2-H signal of the aliphatic chain, along with 4 aromatic signals. This is the only difference to DMT: It shows 1 proton less, as its replaced by a -OH group. In a real spectra this group is invisible due to H-D-exchange in Methanol, as you can see down below and thats the reason why its basically the spectra of DMT with just 1 proton less. Also the NH is not visible due to this reason when measured in a polar-protic solvent, which is the case down below as Methanol was used.
Mestrenova 12 locates the big Methyl-signal very far to high ppm areas and then it hides one of the CH2-units ... Apparently this predicted shift is wrong, indeed its located just in the same area like DMT (see my signature for reference). Strange that it's simulated correctly for DMT and not for Bufotenine as their only difference in structure is very remote from those Methyl-groups, but anyways ... this is irrelevant for the analysis, but now you know why the Methyl-signal seems to be off when comparing simulation and measured sample, but actually the measured sample is correct.

Now here is the measured sample, sadly only 1H-NMR:
Picture of the 1H-NMR-spectra of amorph Bufotenine derived from this TEK. Measured in MeOD at 300 MHz.
Not much to say, all the signals are there (and the big Methyl signal is indeed like DMT of course, not like the wrong simulation). As it is measured in MeOD you can see 2 big solvent peaks. These are marked red, so you can just ignore them. Also there are traces of EA present. The only unidentified peaks are marked gray and these are just traces. In total their integral is just 0,30 and the peak fraction of Bufotenine derived from this number is 97,87 %. Now this does not automatically translate into a purity of 97,87 %, but I may say that still the purity is somewhere between
96 - 99 %. It will be completely pure by > 99,9 % upon 2x recrystallizations in boiling Xylene, as you can see
here at the respective NMR at end of post.
Picture of the GC-Chromatogram of amorph Bufotenine derived from this TEK.
[center]
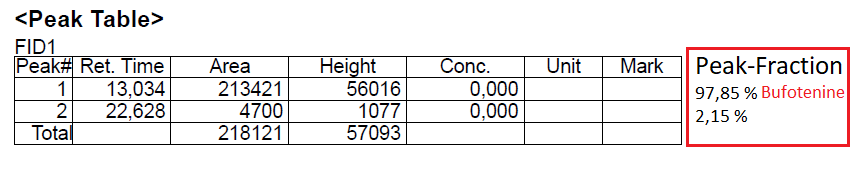
Now when taking a look at the GC-Chromatogram there is practically just 1 peak again. Besides that there is only 1 other peak. Therefore it's very likely that all the foreign peaks from the 1H-NMR are just 1 single other compound. Possibly it's the one that causes the tan coloration and its transfered into the final product as it will still have a very low solubility even in 1:3 EA:Naphtha, so it can't be avoided by that TEK. Still the GC integral is exceptionally high at 97,85 %, so there's nothing to worry.
Looking at these data it seems that even crude Bufotenine derived from this TEK without any additional workup / recrystalization is already practically pure enough for instant usage with
96 % + purity.
Now I guess Bufotenine is open for collaborative research.
